
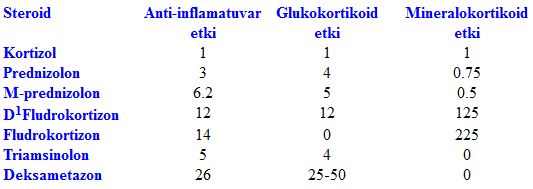
 Adrenal korteks hormonları
Adrenal korteks hormonları
1-Glukokortikoidler (GK); kortizol.
2-Mineralokortikoidler (MK); aldosteron.
3-Androjenler; dihidroepiandrosteron, dihidroepiandrosteron sülfat, androstenedion.
GK-kortizol
ACTH kontrolünde sirkadian ritm. ACTH POMC peptit ailesine dahildir. Hipotalamik CRH ve ADH ile salınım kontrolü sağlanır. Sabah en yüksektir. Hem tip 1 MK hem de GK reseptöre bağlanır. 11BHSD tip 2 kortizona çevirerek inaktive eder ve MK reseptöre bağlanmayı engeller. 11BHSD tip 1 ise kortizonu kortizole çevirerek GK reseptöre bağlanmasını sağlar. %5’i serbesttir.
MK-aldosteron
Başlıca reninle kontrol edilir.Serum Na ve K’a etki eder. ACTH. Sadece tip 1 MK reseptöre bağlanır. Ekstrasellüler sıvıya Na emilimi, renal K ve hidrojen atılımı. %40’ı serbesttir.
Glukokortikoid fonksiyonları
Metabolik: kan glukoz düzeyini düzenler, glukoneogenezi kolaylaştırır ve glikojen sentezini artıtrır. İnsülin düzeyini arttırır. Lipolitik hormonların etkilerini kolaylaştırır. Katabolik ve anti-anabolik etki (yağ hariç). Büyüme hormonu aksını inhibe eder. Gonadal aksı inhibe eder. Mineralo-kortikoid aktivite. Bağ dokusu: kollojen ve bağ doku kaybı yapar. Kalsiyum metabolizması: osteoklast stimülasyonu, osteoblast inhibisyonu yapar. İntestinal Ca emilimini azaltır, PTH salınımını uyarır, idrarla Ca atılımını arttırır fosfat reabsorpsiyonunu inhibe eder. Kardiyovasküler sistem: kardiyak outputu arttırır, vasküler tonusu arttırır, pressör hormonlara duyarlılığı arttırır, sodyum retansiyonunu arttırır. Davranış ve algılama fonksiyonuna etkileri. İmmün sistem: intravasküler lökosit yoğunluğunu arttırır, inflamatuar hücrelerin hasarlı bölgeye göçünü azaltır, immün supresyon yapar.
Adrenal bez hipofonksiyonu
a) Primer adrenal hastalıklar:
Kombine glukokortikoid ve mineralokortikoid eksikliği olanlar
Otoimmün: izole otoimün hastalık (addison hastalığı), tip 1 poliglandüler yetmezlik sendromu, tip 2 poliglandüler yetmezlik sendromu.
İnfeksiyonlar: tüberküloz, mantar, CMV, HIV. Vasküler: bilateral adrenal hemoraji (sepsis, koagülopati, tromboz/emboli, adrenal infarkt).
İnfiltratif: metastatik karsinom/lenfoma, sarkoidoz, amiloidoz, hemokromatoz. Konjenital: konjenital adrenal hiperplazi (21α-hidroksilaz, 3β-HSD eksikliği, 20,22-desmolaz [lipoid hiperplazi]), ACTH’ya yanıt vermeyen adrenal yetersizlik, konjenital adrenal hipoplazi, adrenolökodistrofi, adrenomiyelonöropati.
İyatrojenik: bilateral adrenalektomi, ilaçlar (metirapon, aminoglutetemid trilostane, ketokonazol, o,p’-DDD, RU 486).
İzole mineralokortikoid eksiklik (glukortikoid eksikliği olmayan) (hiperreninemik hipoaldosteronizm)
Kortikosteron metiloksidaz eksikliği, izole zona glomerüloza defekti, heparin tedavisi, ağır hastalıklar, konverting enzim inhibitörleri.
b) Sekonder adrenal hastalıklar
Sekonder adrenal yetersizlik
hipotalamus/hipofiz disfonksiyonu, egzojen glukokortikoidler, ACTH salgılayan tümörlerin çıkarılmasından sonra.
Hiporeninemik hipoaldosteronizm
Diabetik nefropati, tübülointerstisyel hastalık, obstrüktif üropati, otonom nöropati, nonsteroidal anti-inflamatuar ilaçlar, Β-adrenerjik blokerler.
Addison hastalığı
Hem GK hem de MK sekresyonu azalır. Tedavi edilmezse öldürücü olabilir. İzole GK veya MK eksikliklerinde tablo biraz daha hafiftir. Ancak tanı ve tedavi şarttır. Adrenal medulla fonksiyonu genellikle korunur. Tip 1 ve 2 otoimmun poliglanduler sendromun (APS) parçası olabilir. Adrenal yetmezlik gelişimi: sitotoksik T lenfositler tarafından adrenal korteksin destrüksiyonu sonucu oluşur. Hastalığın aktif fazında, yaygın mononükleer hücre infitrasyonu vardır. Adrenokortikal hücrelerde nekroz ve pleomorfizm saptanır. Tip 1 APS (APECED sendromu): autoimmun poli endokrin yetersizlik – candidiasis – ektodermal distrofi. Daha çok çocukluk çağında görülür. Triadı; hipoparatiroidi, adrenal yetersizlik, mukokutanöz candidiasis. Diğer daha az görülen bulguları; hipotiroidi, gonadal yetersizlik (POF), gastrointestinal malabsorbsiyon, tip 1 DM, alopesi areata ve totalis, pernisiyöz anemi, vitiligo, kronik aktif hepatit, diş ve tırnak hipoplazileri, hipofizit, asplenizm, kolelitiyazis. Tip 2 APS (Schmidt sendromu): Triadı; addison hastalığı, otoimmun tiroid hastalıkları (Graves veya Hashimoto), tip 1 DM. Daha çok erişkinlerde görülür. Daha az sıklıkta görülen hastalıklar; pernisiyöz anemi, vitiligo, gonadal yetersizlik, hipofizit, PBS, çölyak hastalığı, myastenia gravis, sjögren sendromu, SLE, parkinson hastalığı. Addisonda belirti ve bulgular: kilo kaybı, yorgunluk, bulantı-kusma, ishal, iştahsızlık, tuz arzulaması, kol ve eklem ağrıları, karın ağrısı,postural baş dönmesi, artmış pigmentasyon (ekstensor yüzlerde, palmar çizgilerde, diş eti mukozasında daha sık).
Addisonda klinik: Semptomlar: halsizlik, kuvvetsizlik, iştahsızlık, bulantı, kusma, konstipasyon, abdominal ağrı, diare, tuz açlığı, baş dönmesi, kas-eklem ağrıları. Bulgular: kilo kaybı, hiperpigmentasyon, hipotansiyon, vitiligo, auriküler kalsifikasyon, hiponatremi, hiperkalsemi, hiperkalemi, azotemi, anemi, eozinofili.
Laboratuvar: hiponatremi, hiperkalemi, hafif metabolik asidoz, azotemi, anemi, hiperkalsemi, lenfositoz, eozinofili, hipoglisemi (özellikle çocuklarda) görülebilir. Akut adrenal yetersizlik (adrenal kriz): tıbbi olarak acil bir durumdur. Tedaviye başlamak için laboratuvar sonuçları beklenmemeli. Hipovolemili kritik hastalarda kortizol, ACTH ve renin için bir plazma örneği alınıp tedaviye başlanmalı. Tedavi; İV bolus tarzında 100 mg hidrokortizon, serum fizyolojik verilir, hipoglisemi varsa %5 dextroz verilir, 3×100 mg hidrokortizon devamında verilir, altta yatan nedenin tedavisi yapılır, replasmana geçiş yapılır.
Adrenal yetersizlik
Tanı: Kronik semptomları olan bir hastada 1 saatlik cosintropin (Synachten, sentetik ACTH) testi uygulanmalıdır. 0.25 mg ACTHİV verilir. 0., 30. ve 60. (İM uygulanmış ise ayrıca 120.) dakikalarda plazma kortizolü ölçülür. Normali herhangi bir zamanda plazma kortizol konsantrasyonunun >20μg/dl olmasıdır.——-Bazal (sabah açlık) plazma kortizolünün <5μg/dL, uyarılmış plazma kortizol konsantrasyonunun <18μg/dL olması aşikar adrenal yetersizliği gösterir. Tedavi gerekir. Bazal plazma kortizolü 10-18 μg/dl arasında olan hastalarda, uyarılmış plazma kortizolü <18μg/dl ise adrenal rezervin yetersizliği vardır. Stres durumları altında kortizol replasmanı yapılır.
Primer-sekonder adrenal yetersizlik ayrımı: Sekonder adrenal yetersizlik;ACTH tarafından adrenal korteksin yetersiz stimulasyonu vardır. Hipotalamo-pitütier-adrenal aks (HPA) boyunca herhangi bir yerdeki lezyon sebep olabilir. Egzojen glukokortikoid kullanımı nedeniyle HPA aksının uzamış supresyonunun bir sekeli olarak görülebilir. Birkaç önemli fark dışında primer adrenal yetersizliğe benzer bulgular vardır. Farklılıklar; hiperpigmentasyon görülmez, sekonder adrenal yetersizlikte MK seviyeleri normal olduğundan tuz arzulama semptomu, hiperkalemi ve metabolik asidoz gibi laboratuvar anormalikleri yoktur. Ancak uygunsuz ADH salınımı sonucu olarak hiponatremi sıktır. ACTH hipofiz hormonlarının en korunmuşudur. Bu nedenle hipofizer lezyona bağlı ACTH eksikliği olan hastalarda diğer ön hipofiz hormon eksiklikleri de aranmalıdır. Primer-sekonder adrenal yetersizlik ayrımı için: sabah açlık plazma ACTH değeri ve 2 saatlik ayakta durma sonrasında plazma renin düzeyine bakılır. Plazma ACTH değeri >20pg/ml (normali: 5-30 pg/ml) ise primer adrenal yetersizlik, <20pg/ml ise sekonder adrenal yetersizlik olasıdır. Ayakta plazma renin düzeyi >3ng/ml/saat ise primer adrenal yetersizlik, <3ng/ml/saat ise sekonder adrenal yetersizlik olasılığı yüksek. Bir saatlik cosintropin testine hem primer hem de sekonder adrenal yetersizlikte yanıt yoktur. Uzun süren sekonder yetersizlikte adrenallerde atrofi vardır.Sekonder adrenal yetersizlik sıklıkla glukokortikoid alımının kesilmesinden sonra görülür.Günaşırı glukokortikoid tedavisi kullanmak, HPA aksını günlük glukokortikoid kullanımına göre daha az suprese eder. İyileşme sırasında ilk olarak ACTH kademeli olarak artar. Bunu plazma kortizol düzeylerinin normalizasyonu, daha sonra ACTH’ya kortizol yanıtının düzelmesi izler. Aksının tam iyileşmesi 1 yılı bulabilir. Burada hız kısıtlayıcı basamak CRH nöronlarının iyileşmesidir.
Tedavi: Yaşam boyu hem glukokortikoidlerle (GK) hem de minerokortikoidlerle (MK) replasman yapılır. GK aşırı tedavi gizli kilo alma ve osteoporoz yapabilir. Tolere edilebilen, minimal kortizol dozu verilmelidir. Sabah 15-20 mg’lık hidrokortizon verilir. GK replasmanı birçok hastada benzerdir.MK replasman dozu bireyseldir. Sentetik MK fludrokortizon’un başlangıç dozu 100μg/gün. Ayakta plazma renini 1-3 ng/ml/saat olacak şekilde doz titrasyonu ayarlanmalıdır.Yeterli dozda GK’e rağmen ayakta plazma renin düzeyi >3ng/ml/saat ise fludrokortizon yetersizdir. Minör bir hastalıkta (bulantı, kusma, 38 OC’den daha düşük bir ateş) hidrokortizon dozu 2 katına çıkarılmalıdır. Oral alamayan hastalarda parenteral hidrokortizon verilmelidir. Majör stresli bir olay (genel anestezi gerektiren cerrahi veya majör travma gibi) geçiren hastalar günlük 150-300 mg’lık parenteral hidrokortizon (3 bölünmüş dozda) almalıdır. İyileşmeyi takiben hızla azaltarak replasman dozuna dönülmelidir. Tüm hastaların tıbbi bilgileri içeren bir künye veya kartı olmalıdır. im hidrokortizon enjeksiyonunu öğrenmelidir. Mineralokortikoidlerin fonksiyonları: renal sodyum ve klor tutulumu, potasyum ve hidrojen atılımı yapar, kan basıncı ve kan hacminin devamlılığını sağlar.
Hiporeninemik hipoaldosteronizm
MK eksikliği renal renin sekresyonununun azalması ile olabilir. AT II düşer ve hiperkalemi ve hiperkloremik metabolik asidozlu hipoaldosteronizm olur. Plazma Na normal olsa da Na deposu sıklıkla yetersizdir. Plazma renin ve aldosteron düzeyleri düşük ve uyaranlara yanıtsızdır. En sık neden DM ve böbreğin tübülointerstisyel hastalığıdır (justaglomerüler aparatusun bozulması). Hiporeninemik hipoaldosteronizmin bir alt tipine otoimmün yetersizlik neden olur. Ayağa kalkma veya volüm kaybı gibi uyaranlara (baroreseptörler aracılığıyla) normal renin yanıtı yoktur. NSAİİ, ACEİ, beta blokerler ile de hipoaldosteronizm gelişebilir. Fludrokortizon ve/veya alfa-1 agonist midodrin hipoaldosteronizme bağlı ortostatik hipotansiyon ve elektrolit anormalliklerini düzeltmede etkilidir.
Hiper-reninemik hipoaldosteronizm
Nadir. Nedenleri; kortikosteron metiloksidazın yetmezliği (aldosteron sentezindeki son basamakta enzim kompleksi),zona glomerulozadaki aldosteron üreten hücrelerin otoimmün harabiyeti, kronik heparin tedavisi.
Konjenital adrenal hiperplazi (KAH)
GK ve MK eksikliğiyle sonuçlanan adrenal steroid sentezindeki bozukluklarıdır. Kortizol sentezindeki yetersizlikten dolayı ACTH artar. Takiben adrenal hiperplazi ve enzim blokajından önceki steroidlerin aşırı üretimi vardır. Beş major KAH tipi vardır. Her bozukluğun klinik tablosu eksik ve fazla olan steroide bağlı. Tümü otozomal resesif geçişlidir.
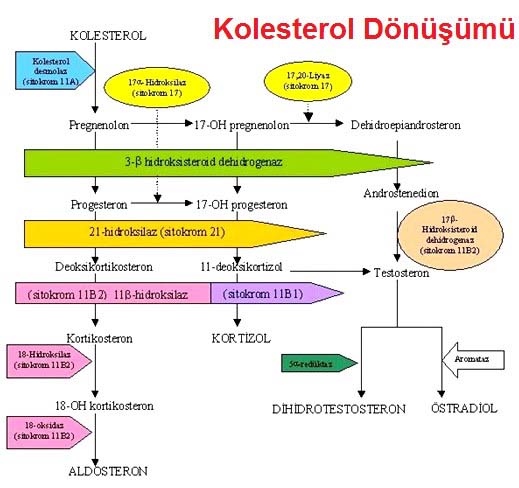
21-Hidroksilaz eksikliği
En sık tip (%95). 17-OH P ve P’nun, sırasıyla 11-deoksikortizol ve deoksikortikosterona dönüşümü yoktur. Sonuçta kortizol ve aldosteron üretimi eksiktir. Kortizol eksikliği ACTH salınımına yol açar. Bu da 17-OH progesteron ve progesteronun aşırı üretimine neden olur. Artmış ACTH üretimi, testosterona dönüştürülebilen androstenedion ve DHEA sentezinin artışına neden olur. İki klinik fenotipi vardır; 1) Klasik 21-hidroksilaz eksikliği; genellikle doğumda veya çocukluk çağında teşhis, 2) Geç başlangıçlı 21-hidroksilaz eksikliği; puberte sırasında veya sonrasında ortaya çıkar. Klasik tipte hastaların 2/3’sinde değişik derecelerde MK eksikliği vardır; tuz kaybettiren formdur. 1/3 ünde tuz kaybı yoktur; basit virilizan tiptir. Hem aldosteron üretimindeki azalma, hem de MK antagonisti prekürsörlerin (P ve 17-OH P) artışı ile ağır enzimatik blokaj olan olgularda tuz kaybına katkıda bulunur. Tanıda plazma 17-OH progesteron düzeyinin ölçümü önemlidir; >2 ng/ml tanıyı koydurur. Geç başlangıçlı 21-hidroksilaz eksikliği tipinde bir varyant alleli bulunduğu için hafif tipte enzim defekti bulunur.İnsanlarda en sık görülen otozomal resesif hastalıktır.Pubertal virilizasyon bulguları (hirsutizm ve akne) ve amenore veya oligomenore ile karakterizedir. Açıklanamayan hirsutizm ve adet bozukluğu olan kadınlarda bu sendrom düşünülmelidir. Tanı; 250 µg.lık sentetik ACTH (iv formu) verilmesinden 30 dk. sonra plazma 17-OH P düzeyinin belirgin yükselmesi (>15 ng/ml olması) ile tanı konur. Klasik tipte 21-hidroksilaz eksikliğinin tedavi amacı; GK ve MK replasmanı, ACTH ve androjen aşırı üretiminin supresyonu ve çocuklarda normal gelişim ve seksüel olgunlaşmayı sağlamaktır. Basit virilizan formdakiler dahil tüm etkilenmiş hastalara fizyolojik dozda hidrokortizon ve fludrokortizon replasmanı yapılır. Fazla androjenlerin zararlı etkileri antiandrojen bir ajan (flutamid gibi) ve bir aromataz inhibitörünün (testolakton; testesteronun östrojene dönüşümünü bloke eden) ilaçların kullanımı ile önlenebilir. Geç başlangıçlı 21 hidroksilaz eksikliği için geleneksel tedavi dexametazon (0.5mg/gün). Spironolakton (100-200mg/gün) gibi bir antiandrojenin kullanımı muhtemelen daha etkilidir ve daha az yan etkisi vardır. Geç başlangıçlı tipte MK replasmanına gerek yoktur.
11 hidroksilaz (CYP11B1) eksikliği
KAH’lı vakaların yaklaşık %5’ini oluşturur. Bu sendromda 11 deoksikortizolün kortizole ve 11 deoksikortikosteronun kortikosterona (aldosteron prekürsörü) dönüşümü bloke olur. Hastalarda genellikle HT ve hipokalemi vardır. 21 hidroksilaz eksikliğindeki gibi virilizasyon görülür. Bazal yada ACTH sitimülasyondan sonra 11 deoksikortizol düzeyindeki artış ile tanı konur.
KAH’ın nadir formları
3-ßOH steroid dehidrogenaz (3-ß-HSD), 17 hidroksilaz (CYP17) ve steroidojenik akut regülatuar (StAR) protein eksiklikleridir.
Adrenal bez hiperfonksiyonu
a) Kortizol aşırı salınımı; Cushing sendromu.
b) Aldosteron fazla salınımı; primer aldosteronizm (Conn sendromu), sekonder aldosteronizm.
c) Androjenlerin fazla salınımı; konjenital adrenal hiperplazi (KAH), adrenokortikal kanser.
Cushing sendromu (CS)
Etyoloji: 1) Steroidlerin fazla kullanılması. 2) Aşırı ACTH salınması (ACTH bağımlı) (%85); hipofiz adenomu (cushing hastalığı) (%80), tömörlerin ektopik ACTH salgılaması (%5) (bronşiyal karsinoid, timik karsinoid, adacık h. Tm, ac küçük hücreli ca.), ektopik CRH. 3) Aşırı kortizol salınması (ACTH bağımsız) (%15); adrenal adenom, adrenal neoplazi, mikronodüler ve makronodüler adrenal hiperplazi.
Ayırıcı tanı: Fizyolojik hiperkortizolemiler; stres, gebeliğin 3.trimesteri ve düzenli ağır egzersiz. Diğer patolojik durumlar; depresyon, alkolizm, anoreksiya nervoza, panik bozukluk ve alkol veya narkotikten kesilme. Obezite.
Belirti ve bulgular: yağ dağılımında değişiklik, adet düzensizliği, ince ve pletorik cilt, yüzde yuvarlaklaşma, iştah artışı, uykusuzluk, kan basıncında artış, hiperlipidemi (kolesterol, TG), mental değişiklikler (öfori, konsantrasyon ve hafızada azalma), glukoz tolerans bozukluğu, DM, stria, hirsutismus, proksimal kas güçsüzlüğü, psikolojik bozukluklar (emosyonel labilite, depresyon, mani, psikoz), azalmış libido, impotans, akne, osteoporoz, fraktür, virilizasyon, kolay berelenme, yara iyileşmesinde gecikme, ödem, infeksiyona yatkınlık, katarakt, buffalo hump.
Laboratuvar bulguları: Glukoz intoleransı/hiperglisemi ve glukozüri, serum alkalen fosfataz yüksekliği, granülositoz, trombositoz, hiperkolesterolemi, hipertrigliseridemi. Alkaloz ve düşük potasyum ektopik ACTH olgularında.
Tanı: 1-Hiperkortizoleminin tanısı: deksametazon supresyon testi,24 saatlik idrar kortizolü,diürinal kortizol ritmi, tükrük kortizolü. 2-Etyolojik tanı: ACTH ölçümü, 2 günlük deksametazon supresyon testi, yüksek doz deksametazon testi, CRH ve İPSÖ, adrenal, hipofiz ve ektopik odağı görüntüleme yöntemleri.——-Cushing sendromu tanısı koyabilmek için en az iki ilk basamak testin bozuk olması gerekir. İlk basamakta yer alan testler gece yarısı tükrük kortizolü, idrar kortizolü ve düşük doz dekzametazon baskılama testidir. İdrar ve tükrük kortizol ölçümleri en az iki kez yapılmalıdır. İdrar kortizol ekskresyonu belirgin yüksek olmalıdır (normal üst sınırın en az üç misli) veya Cushing sendromu tanısı kesin değilse diğer testler yapılmalıdır.
Tedavi: Cerrahi: TNTS adenomektomi,unilateral adrenalektomi, ektopik odağın çıkarılması. RT: gamma knife. Medikal: ketokonazol, mitotan, metirapon, aminoglutetimid, mifepriston ve trilostan.
Mineralokortikoid fazlalığı
1-Primer aldosteronizm: aldosteron sekrete eden adenom (%75), bilateral adrenal hiperplazi (%25), aldosteron sekrete eden karsinom (%1),glukokortikoitle baskılanabilen hiperaldosteronizm. 2-Adrenal enzim eksiklikleri: 11 ß-OH’laz eksikliği, 17 α-OH’laz eksikliği, 11 β-HSD eksikliği (görünürde mineralokortikoid fazlalığı sendromu). 3-Ekzojen mineralokortikoitler: likoris, karbenoksolon, fludrokortizon (11 β-HSD inhibisyonu). 4-Sekonder hiperaldosteronizm: Hipertansiyonla birlikte olanlar; akselere hipertansiyon, renovasküler hipertansiyon, östrojen tedavisi, renin sekrete eden tümörler. Hipertansiyonla birlikte olmayanlar; bartter sendromu, Na kaybettiren nefropati, renal tübüler asidoz, diüretik/laksatif kullanımı, ödemli durumlar (siroz, nefroz, konjestif kalp yetmezliği).
Primer hiperaldosteronizmde MK aktivitesindeki artma; tuz tutulumu, hipertansiyon, hipokalemi ve metabolik alkaloz vardır. Genellikle HT veya hipokaleminin değerlendirilmesi sırasında tanınır.Kür olabilen HT formunu oluşturur. HT %2’sinden daha azında primer aldosteronizm vardır. Genellikle 30-50 yaşta görülür. Kadın/erkek oranı: 2/1’dir.
Belirti ve Bulgular: MK aktivitesinde artış ile; Tuz tutulumu, Hipertansiyon, Hipokalemi, Metabolik alkaloz görülür. Hipokalemiye bağlı; halsizlik, kas güçsüzlüğü, noktüri, bitkinlik ve baş ağrısı görülür. Ağır hipokalemide; polidipsi, poliüri, paresteziler ve hatta intermittan paralizi ve tetani vardır. Metabolik alkaloza bağlı Trousseau veya Chovostek pozitifliği görülür.
Tanı: HT tanısı olan hastalarda K ölçümü şarttır. Kullanılan diüretik ve RAA sistemini etkileyen anti-hipertansifler kesilir. Yeterli tuz alımı. Ayakta plazma aldosteron ve renin aktivitesi ölçümü yapılır. Serum aldosteron/PRA oranı > 30ng/dL/ng/mL/hr olması ve serum aldosteron düzeyi >15ng/dL olması hiperaldosteronizm tanısını düşündürür.
Tedavi: Tümörü lokalize etmek için adrenal BT çekilir. Eğer bir adrenal bezde adenom, diğer bez normal, biyokimyasal test sonuçları adenom ile uyumlu ise; tek taraflı adrenalektomi yapılır. Biyokimyasal bulgular adenom ile uyumlu, ancak BT sonuçları bilateral hiperplazi ile uyumlu ise aldosteron ve kortizol ölçümleri için adrenal venöz sampling/iyodokolesterol sintigrafisi çekilir. Biyokimyasal ve lokalizasyon bulguları bilateral hiperplazi ile uyumlu olan hastalarda medikal tedavi (genellikle spironolakton) yapılır.
Biyokimyasal sonuçları bilateral hiperplazi ile uyumlu olan hastalarda deksametazonla baskılanabilen hiperaldosteronizm olabilir. Deksametazon testi yapılır. Nadir otozomal dominant bir durumdur. Deksametazon ile hiperaldosteronizm düzelir. Bu hastalıkta erken yaşta hemorajik inme görülebilir.
Sekonder hiperaldosteronizm
RAAS aktivasyonuna sekonder olarak hiperaldosteronizm ve HT vardır.Akselere (hızlanmış) HT’lu hastalar. Vasküler hipertansiyonlarda, östrojen tedavisi alanlarda, nadiren renin salgılayan tümörlü hastalarda görülür.
Hipertansiyonsuz sekonder hiperaldosteronizm: Bartter sendromu, sodyum kaybettiren nefropatiler, renal tübüler asidoz, diüretik veya laksatif alışkanlığı olanlarda görülür. Bartter sendromunda primer renal tübüler NaCl emilim bozukluğu sonucu Na-K-H pompası fazla çalışır. Diüretik kullanımına benzer bir tablo oluşturur. Hipertansiyon yoktur.
Adrenal androjenlerin fonksiyonları: DHEA, DHEAS, AS. Periferde testosteron ve DHT’a dönüşür. Erkekte androjenik duruma katkı %5, kadında folliküler fazda %70, siklus ortasında %40. Libido sağlar.
Adrenal medulla hormonları
1.Epinefrin. 2. Norepinefrin. 3. Dopamin.
Fizyoloji: Adrenal medulladan üretilen major katekolamin norepinefrindir; α agonist etki baskındır, vazokonstrüksiyona neden olur. Epinefrin başlıca β reseptörler üzerine etki eder; kalp üzerinde (+) inotropik ve kronotropik etkiler, periferik damarlarda vazodilatasyon yapar ve hipoglisemiye yanıt olarak plazma glukoz konsantrasyonunu artırır. Dolaşan dopaminin etkisi tam bilinmemektedir. Norepinefrin santral sinir sistemi ve sempatik post ganglionik nöronlarda da üretilebilir. Epinefrin yalnızca adrenal medullada üretilir. Adrenal medullanın norepinefrin salınımına katkısı nispeten düşüktür. Bilateral adrenalektomi dolaşımdaki norepinefrin düzeylerinde minimal değişiklilere neden olur, epinefrin düzeyleri belirgin olarak azalır. Medullanın hipofonksiyonu hafif fizyolojik etki gösterirken, katekolaminlerin hiperfonksiyonu feokromositoma denen klinik sendroma neden olur.
Feokromasitoma
Vücutta herhangi bir sempatik gangliondan kaynaklanabilir.%90ından daha fazlası adrenal medulladan kaynaklanır. Ekstra adrenal tümörlerin çoğunluğu mediastende veya batındadır. Olguların ancak %5’inde bilateraldir görülür ve bunlar familyal sendromların bir parçası olabilir. MEN-2A veya 2B’nin bir parçası olabilir. Tip-2A (Sipple sendromu); tiroidin meduller kanseri, hiperparatiroidi ve feokromositoma. Tip-2B ise meduller tiroid kanseri, mukozada nöromlar, intestinal ganglionöromlar, marfanoid görünüm ve feokromositoma vardır. Feokromositomalar nörofibramatozis, serebelloretinal hemonjioblastozis (von Hippel-Lindau sendromu) ve tuberoz siklerozisle birlikte olabilir.
Klinik: feokromositomalarda çoğunlukla norepinefrin salgılanır ve bu nedenle en sık bulgu HT (sıklıkla paroksismal), baş ağrısı, çarpıntı, terleme, flushing (özellikle yüzde kızarıklık), anksiyete, bulantı, halsizlik, kilo kaybı, karın ve gögüs ağrıları. Semptomlar emosyonel stres, egzersiz, anestezi, abdominal basınç ve tirozin içeren gıdaların alınması ile ortaya çıkar. Ortostatik hipotansiyon görülebilir. Kan basıncında geniş dalgalanmalar karakteristiktir. Feokromositoma ile ilişkili hipertansiyon genellikle standart antihipertansif tedaviye iyi yanıt vermez.
Tanı: plazma serbest metanefrin ve normetanefrin düzeylerinin ölçülmesi en değerli testtir. Plazma serbest metanefrin düzeyinin > 0.61 nmol/L ve serbest normetanefrin düzeyinin > 0.31 nmol/L olması FEO tanısı ile uyumludur. Eğer değerler hafifçe yüksek ise clonidin supresyon testi yapılır. Klonidin 300 mg ağızdan verilir ve plazma katekolaminleri (serbest metanefrin ve normetanefrin dahil) önce ve 3 saat sonra ölçülür. Normal bireylerde katekolamin düzeyleri normal orana dönerken feokromositomada bu düzeyler değişmez veya artar.
FEO tanısında biyokimyasal testler: Plazma normetanefrin ve metanefrin, plazma norepinefrin ve epinefrin, idrar normetanefrin ve metanefrin, idrar norepinefrin ve epinefrin, idrar VMA.
Biyokimyasal tanı sonrası adrenal BT çekilir. Birçok adrenal feokromositoma bu tarama sırasında kolaylıkla görülür. Eğer BT tarama (-) ise ekstra adrenal feokromositomalar iyot 131-metaiodobenzilguanidin (¹³¹І-MİBG), octreotid sintigrafisi veya abdominal MRI ile lokalize edilebilir.
Tedavi: Eğer lezyon lokalize edilebilirse seçilecek tedavi yöntemi cerrahidir. Cerrahi sırasında önemli komplikasyonlar (ağır hipertansiyon veya hipotansiyon) görülebildiğinden cerrahi öncesi 1-2 hafta süre ile hastalarda fenoksibenzminle α blokaj yapılmalıdır. β adrenerjik blokerler cerrahi öncesinde veya sırasında kullanılabilir (ancak α bloker uygulanmadan β bloker verilmesi hipertansif atağı şiddetlendirir). FEO’ların yaklaşık %5-10’u maligndir. ¹³¹I-MIBG veya KT yararlı olabilir. Prognoz genellikle kötüdür. Tümörden katekolamin salınımını azaltmak için α-metil-para-tirozin (katekolamin sentezinde hız kısıtlayıcı enzim) kullanılabilir.
YAZIYI PAYLAŞ
Yazar Hakkında
Dr. Enes Başak
Aile Hekimliği
 Sürrenal hastalıkların ayırıcı tanısı
Sürrenal hastalıkların ayırıcı tanısı Kortizol (stres hormonu) nedir? Ne işe yarar? Yüksekliği ve düşüklüğü
Kortizol (stres hormonu) nedir? Ne işe yarar? Yüksekliği ve düşüklüğü Endokrin acillerin tanı ve tedavi yöntemleri
Endokrin acillerin tanı ve tedavi yöntemleri Cushing sendromu nedir? Nedenleri, belirtileri ve tedavisi
Cushing sendromu nedir? Nedenleri, belirtileri ve tedavisi Prof. Dr. Kaynar: İmmün plazma, Kovid-19’a karşı en önemli tedavilerden birisi
Prof. Dr. Kaynar: İmmün plazma, Kovid-19’a karşı en önemli tedavilerden birisi
YORUMUNUZ VAR MI?