
Genetik hastalıklar: Huntington
 Huntington (HD) veya Chorea Huntington hastalığı çok nadir görülmesine rağmen çok ciddi ve günümüzde henüz tedavisi olmayan kalıtsal bir beyin hastalığıdır. Hastalığa 4. Kromozomda bulunan HTT adındaki gende meydana gelen bir mutasyon sebep olur. Hastalığın ilk belirtileri genellikle 35 ila 45 yaşları arasında başlar, ancak erken çocukluk döneminde veya sadece yaşlılıkta da ortaya çıkan formları da vardır. Bu farklılık, hastalığa neden olan mutasyonun genomun neresinde bulunduğuna ve tekrar eden sekansın uzunluğuna bağlı olarak değişir. Gevşek kas tonusu, koordine edilemeyen istemsiz hareketler, kol–bacak–gövde ve yüz kaslarında ani ve istemsiz kasılmalar hastalığın tipik belirtileridir.
Huntington (HD) veya Chorea Huntington hastalığı çok nadir görülmesine rağmen çok ciddi ve günümüzde henüz tedavisi olmayan kalıtsal bir beyin hastalığıdır. Hastalığa 4. Kromozomda bulunan HTT adındaki gende meydana gelen bir mutasyon sebep olur. Hastalığın ilk belirtileri genellikle 35 ila 45 yaşları arasında başlar, ancak erken çocukluk döneminde veya sadece yaşlılıkta da ortaya çıkan formları da vardır. Bu farklılık, hastalığa neden olan mutasyonun genomun neresinde bulunduğuna ve tekrar eden sekansın uzunluğuna bağlı olarak değişir. Gevşek kas tonusu, koordine edilemeyen istemsiz hareketler, kol–bacak–gövde ve yüz kaslarında ani ve istemsiz kasılmalar hastalığın tipik belirtileridir.
 Huntington (HD) veya Chorea Huntington hastalığı çok nadir görülmesine rağmen çok ciddi ve günümüzde henüz tedavisi olmayan kalıtsal bir beyin hastalığıdır. Hastalığa 4. Kromozomda bulunan HTT adındaki gende meydana gelen bir mutasyon sebep olur. Hastalığın ilk belirtileri genellikle 35 ila 45 yaşları arasında başlar, ancak erken çocukluk döneminde veya sadece yaşlılıkta da ortaya çıkan formları da vardır. Bu farklılık, hastalığa neden olan mutasyonun genomun neresinde bulunduğuna ve tekrar eden sekansın uzunluğuna bağlı olarak değişir. Gevşek kas tonusu, koordine edilemeyen istemsiz hareketler, kol–bacak–gövde ve yüz kaslarında ani ve istemsiz kasılmalar hastalığın tipik belirtileridir.
Huntington (HD) veya Chorea Huntington hastalığı çok nadir görülmesine rağmen çok ciddi ve günümüzde henüz tedavisi olmayan kalıtsal bir beyin hastalığıdır. Hastalığa 4. Kromozomda bulunan HTT adındaki gende meydana gelen bir mutasyon sebep olur. Hastalığın ilk belirtileri genellikle 35 ila 45 yaşları arasında başlar, ancak erken çocukluk döneminde veya sadece yaşlılıkta da ortaya çıkan formları da vardır. Bu farklılık, hastalığa neden olan mutasyonun genomun neresinde bulunduğuna ve tekrar eden sekansın uzunluğuna bağlı olarak değişir. Gevşek kas tonusu, koordine edilemeyen istemsiz hareketler, kol–bacak–gövde ve yüz kaslarında ani ve istemsiz kasılmalar hastalığın tipik belirtileridir.Hastalığın etkilerini engelleme yönünde yapılan müdahalelerin etkileri sınırlıdır. Sinir hücrelerini korumak ve ileride oluşabilecek nöronal dejenerasyonu durdurabilmek için çok sayıda madde test edilmesine rağmen şu ana kadar bu maddelerin hiçbiri hastalığın seyri üzerinde önemli bir etkiye sahip olamadı. Bununla birlikte, Huntington hastalığının semptomlarını azaltmaya yardımcı olabilecek bazı ilaçlar bulunmaktadır.
Teşhis
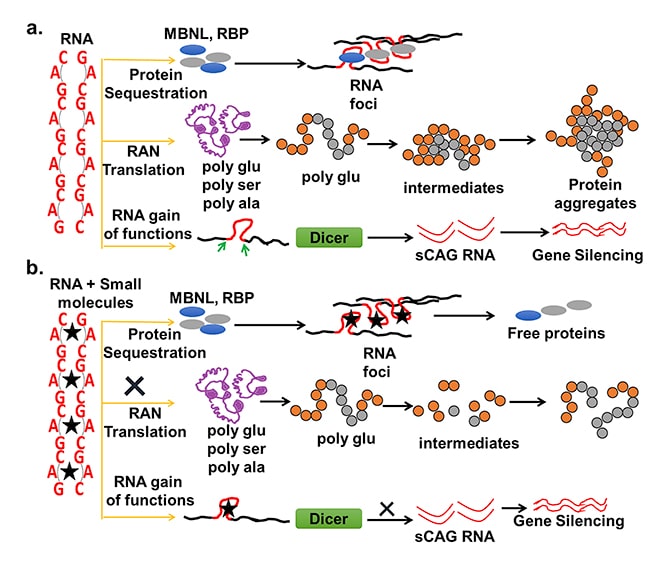 Hastalık genetik bir test ile ortaya çıkartılır. Hasta ve yakınları için genetik testin sonuçlarını beklemek endişe verici ve zor bir dönemdir. Test sonuçları çoğu zaman siya-beyaz şeklinde kesin olmakla beraber küçük bir azınlık Gri Alanda (Azaltılmış penetrasyon) yer alır. Bu tür sonuçların ne anlama geldiğini hem anlamak, hem de hastaya anlatmak oldukça zordur.
Hastalık genetik bir test ile ortaya çıkartılır. Hasta ve yakınları için genetik testin sonuçlarını beklemek endişe verici ve zor bir dönemdir. Test sonuçları çoğu zaman siya-beyaz şeklinde kesin olmakla beraber küçük bir azınlık Gri Alanda (Azaltılmış penetrasyon) yer alır. Bu tür sonuçların ne anlama geldiğini hem anlamak, hem de hastaya anlatmak oldukça zordur.
Bu yazımızda hem hastalık hakkında genel bir bilgi, hem de gri alan yer alanda bazı durumlara açıklık getirilmeye çalışılacaktır. Makalenin sonunda ise geliştirilmekte olan ve oldukça umut vaad eden etkili bir ilaçlı tedaviden bahsedilecektir.
Hastalığın genetik özelliği
Genler, DNA denilen genetik materyalden oluşur. DNA, tüm yaşamın şifresidir ve bu şifre A, C, G ve T olmak üzere dört harften oluşur. Bu harflerin bilimsel adı Nükleotit’dir. 4. Kromozomda HTT, ya da diğer adı HK olan bir gen bulunur. Bu genin her insanda bir anneden bir de babadan olmak üzere iki kopyası bulunur.
Kromozomal hastalıklar ve gebelik: 35 yaş üstü hamilelikler neden riskli
HTT geni Huntington adında bir protein kodlar ve HTT geninde meydana gelen bir mutasyon bu proteinin hatalı kodlanmasına ve buna bağlı olarakta Huntington hastalığının ortaya çıkmasına sebep olur. Mutasyon, genin başlangıç kısmına yakın bir bölgesinde „CAG, CAG, CAG, CAG, CAG, CAG, CAG….CAG“ şeklinde arka arkaya defalarca tekrarlanan bir nükleotit dizilimi şeklindedir.
HTT genindeki CAG tekrar sayısı kişiden kişiye değişebilmekte ve bu sayı bazılarında 15, bazılarında ise 120’ye kadar çıkabilmektedir. Sağlıklı bir insanda ortalama tekrar sayısı yaklaşık 17’dir. Sağlığımız ve ömrümüz bu “CAG” diziliminin tekrar sayısına bağlı olarak değişmektedir. Huntington, dominant bir hastalıktır. Bu şu anlama geliyor: HTT geninin iki kopyasından birisinde yüksek sayıda CAG bulunması durumunda hasta olmak kaçınılmazdır.
Güvenli sayı nedir
Eğer HTT geninin her iki kopyasındaki CAG sayısı 26 veya daha az tekrara sahipse o kişi ve çocukları sağlıklı sayılır. HTT geninin bir kopyası 40 veya daha fazla sayıda CAG dizilimine sahip ise bu, o kişilerin yaşamlarının bir noktasında Huntington hastalığı ile yüz yüze gelecekleri anlamına gelmektedir. Ayrıca bu durumda bunların çocukları da % 50′ oranında risk altındadır.
Gri alan
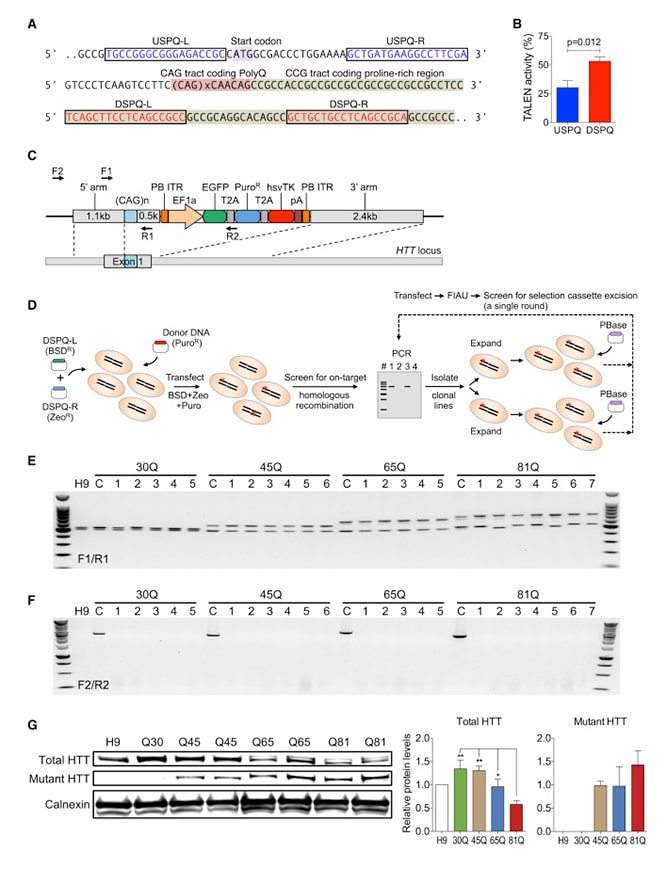 CAG tekrarlarının sayısı 27 ile 39 arasında olması genellikle Gri alan olarak tanımlanır ve gri alanda bulunanların test sonuçlarını tanımlamak hem oldukça karmaşık, hem de bunu hastaya anlatmak bir hayli zordur. Örneğin; 36 ila 39 CAG tekrar sayısına sahip kişilerin bazılarında hastalık belirtileri ortaya çıkarken bazılarında çıkmamaktadır. Bu yüzden bu aralıkta bulunan kişiler için hastalığın geleceği konusunda bir öngörüde bulunmak zordur ama eğer semptomlar yaşamın herhangi bir döneminde belirmeye başladıysa, daha sonraki bir dönemde hastalık başlar, ama şiddeti azdır. Bu forma sahip bireylerin çocukları ise yaklaşık % 50 oranında risk altındadır.
CAG tekrarlarının sayısı 27 ile 39 arasında olması genellikle Gri alan olarak tanımlanır ve gri alanda bulunanların test sonuçlarını tanımlamak hem oldukça karmaşık, hem de bunu hastaya anlatmak bir hayli zordur. Örneğin; 36 ila 39 CAG tekrar sayısına sahip kişilerin bazılarında hastalık belirtileri ortaya çıkarken bazılarında çıkmamaktadır. Bu yüzden bu aralıkta bulunan kişiler için hastalığın geleceği konusunda bir öngörüde bulunmak zordur ama eğer semptomlar yaşamın herhangi bir döneminde belirmeye başladıysa, daha sonraki bir dönemde hastalık başlar, ama şiddeti azdır. Bu forma sahip bireylerin çocukları ise yaklaşık % 50 oranında risk altındadır.
Başka bir gri alan da 27 ile 35 tekrara sahip olanlardadır. Bu tekrara sahip kişilerin HTT geninin bir kopyası sağlıklı ise bu kişilerde Huntington görülmez ancak çocukları % 50 risk altındadır.
Gelecek nesillerde durum
HTT geninde mutasyon bulunan bireyin çocukları ve torunlarında CAG tekrar sayısı kararsız duruma geçebilmektedir. Yani yeni nesillerde CAG tekrar sayısı artabileceği gibi azalabilmektedir de… HTT genindeki bu dengesizlik doğal olarak gelecek nesiller hakkında bir tahmin yürütmeyi dezorlaştırıyor. HTT geninin neden dengesiz olduğu tam olarak bilinmese de sorunun DNA’nın kopyalanması esnasında oluşan hatalardan kaynaklandığı tahmin ediliyor.
Aile öyküsü önemli mi?
Huntington hastalığına yakalananların yaklaşık %10’nunda aile öyküsünün olmadığı görülse de, bu istatiksel bilgilerin güvenilir olduğunu söylemek pek doğru sayılmaz, Bu yüzden aile öyküsünün olmaması diğer aile bireylerinde ve bunların doğacak çocuklarda hastalığın görülmeyeceği anlamına gelmiyor.
İstatistikleri yanıltan birkaç sebep var
- Özellikle geçmiş yıllarda güvenilir bir genetik test olmadığı için hastalığa Huntington yerine Parkinson veya başka bir nörodejeneratif hastalık teşhisi konulması (Hatta bu hatanın günümüz de bile yapılması mümkün.)
- İstatiksel verileri yanıltan başka bir konu da potansiyel olarak Huntington riski taşıyan bireyin ilk belirtiler ortaya çıkmadan başka bir hastalıktan ölmesi.
- Yine istatistikleri yanıltan bir başka konu daha var o da genetik mutasyonun durumu ile ilgili. Mesela orta uzunlukta CAG tekrar sayısına sahip bir bireyin kendisi hasta olmasa da çocuklarında bu risk her zaman var. Yapılan genetik çalışmalar çocuklarında Huntington görülen ebevyenlerde genellikle orta uzunlukta, yani 27 ile 35 CAG tekrar sayısına sahip olduğunu gösteriyor.
Parkinson hastalığı neden olur? Belirtileri, teşhisi ve evreleri
Özet
Dördüncü kromozomun kısa kolunda bulunan HTT genide CAG şeklinde bir üçlü dizilim var. CAG, üç nükleotitin kısa yazılmış halidir. Buna göre CAG, 1 adet Cytosin (C), 1 adet Adenin (A), 1 adet Guanin (G) den oluşmaktadır. Huntington hastalığı bu dizilimin ard arda tekrar sayısı ile ilintilidir. Başka bir ifade ile bu sayı sağlıklı kişilerde az, Huntington hastalarında fazladır.
CAG sayısı dört kategoriye değerlendirilmektedir.
 25 ve daha az tekrar sayısına sahip olanlarda hastalık oluşmaz. Bu aralık normal kabul ediliyor.
25 ve daha az tekrar sayısına sahip olanlarda hastalık oluşmaz. Bu aralık normal kabul ediliyor.- 26-36 arası Gri alandır. Bu aralıkta bulunanlar büyük bir olasılıkla Huntington hastalığına yakalanmayacaklar ancak bu kişilerin yumurta veya sperm hücreleri daha yüksek sayıda CAG sahip olabilir. Bu da gelecek nesilde yeni mutasyonların oluşmasına sebep olabilmektedir. Bu nedenle bu kişilerin çocukları için bir risk olduğunu söylemek yanlış olamaz.
- 36-39 tekrar sayısına sahip olanların bazılarında hastalık belirtileri görülürken, bazılarında görülmez. Bu gruptakilerde eğer hastalık günün birinde ortaya çıkacaksa, bu oldukça geç yaşta ve hafif ilerlerleyen bir formda ortaya çıkacaktır. Bu nedenle bu gruptakilerde çoğu vaka teşhis edilemez.
- 40 tekrar sayısı pozitiftir. Yani hastalık yaşamın bir döneminde mutlaka ortaya çıkar.
40 tekrar ve üzerindekilerde ölüm yaşı
- 40 tekrarda ortalama ölüm yaşı 59 dur.
- 41 tekrarda ölüm yaşı 54,
- 42 tekrarda ölüm yaşı 37,
- 50 tekrarda ölüm yaşı 27,
Bu değerler her ne kadar CAG sayısı ile hastalığın başlangıcı ve hastalığın seyri ve ölüm yaşı konusunda bir şey söylese de bu genel bir tanımdır. Bireysel durum kimi zaman farklı olabilmekedir. Bazen aynı CAG değerine sahip iki bireyde hastalığın ortaya çıkma yaşı farlı olabilmektedir. Hatta hasta olan ikizlerde bile hastalığın ortaya çıkma yaşı farklı farklı olabilmektedir.
Özellikle 50’nin altındaki CAG değerleri için hastalığın ortaya çıkma yaşı kişiden kişiye on yıl varan farklılıklar gösterebilmektedir. Ayrıca mutasyona ek olarak, diğer genler ve çevresel faktörler de rol oynadığı tahmin ediliyor.
Tedavide umut
Yukarıda belirtildiği gibi CAG sayısında meydana gelen anormal artışın Huntington proteininde artışa, bu da sinirler üzerinde patojen etki yaparak hastalığın ortaya çıkmasına sebep oluyor. Aslında başka genler üzerinde benzer sebepler kaynaklanan başka genetikler hastalıklar da var. Bu tür hastalıkların tedavisinde değişik stratejiler uygulanıyor.
Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
Bu uygulamalardan biri klasik bir yöntem. Bu tedavi yöneminde genlerin ürünü olan proteinler hedef alınıyor. Buna göre eğer hastalık eksik proteinden kaynaklanıyorsa, protein ilavesi, fazla proteinden kaynaklanıyorsa, fazla proteini bloke etme şeklinde oluyor.
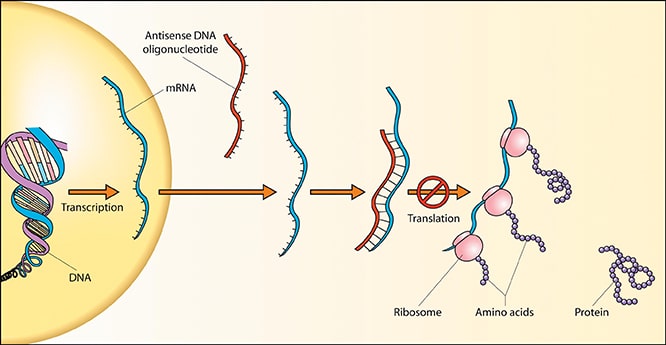
Yeni yöntem: Hedefe kilitlenen Anti-sens ilaçlar
Bilimsel adı Antisense-Oligonukleotide olan Antisens ilaçlar, hastalığa neden olan mutasyona uygun “anti sekanslar” şekilde hazırlanıyor. Huntington hastalığı için geliştirilen Antisens ilaçlar fazla olan CAG dizilimini bloke ederek hastalığı tedavi ediyor. Yani karşıt dizilim fazla olan CAG dizilimine yapışarak gereksiz protein üretimini durduruyor.
Klinik çalışmalar devam ediyor
Yukarıda daha kolay anlaşılması için çok fazla ayrıntıya girilmeden anlatılmaya çalışılan ilaçlar üzerinde birçok firma çalışma yapıyor. Klinik öncesi aşamada olan bu ilaçların patenti büyük ilaç firmaları tarafından çoktan alındı. Antisens ilaçların klinik öncesi yapılan çalışmalarının başarılı olduğunu belirtiliyor.
Benzer konuda hazırlanmış diğer makaleler
- Sporun Alzheimer’a karşı koruyucu etkisi
- Kokuların anıları çağrıştırması ve koku ile Alzheimer arasında ilişki
- Köpek balığı, Alzheimer ve Parkinson tedavisinde bir umut olabilir
- Alzheimer’a karşı alınacak yedi basit önlem
- Epilepsi hastaları için yeni bir umut: Gen terapisi
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics of the University of Cologne
++++++++++++++++++++++++
Kaynaklar
Late onset of Huntington's disease
Length of Uninterrupted CAG, Independent of Polyglutamine Size, Results in Increased
Rationally designed small molecules targeting toxic CAG repeat RNA that causes Huntington's disease (HD) and spinocerebellar ataxia (SCAs)
Unbiased Profiling of Isogenic Huntington Disease hPSC-Derived CNS and Peripheral Cells Reveals Strong Cell-Type Specificity of CAG Length Effects.
Somatic mosaicism in sperm is associated with intergenerational (CAG)n changes in Huntington's disease.
Mortality rate of Huntington disease in Japan: Secular trends, marital status, and geographical variations
The prevalence of Huntington's chorea in South Africa..
Huntington's disease in black kindreds in South Carolina.
DNA haplotype analyses of Huntington disease reveals clues to the origins and mechanisms of CAG expansion and reasons for geographic variations of prevalence.
Ancestral differences in the distribution of the D2642 glutamic acid polymorphism is associated with varying CAG repeat lengths on normal chromosomes: insights into the genetic evolution of Huntington disease.
Reproductive options for prospective parents in families with Huntington's disease: clinical, psychological and ethical reflections
IONIS-HTTRx Shows Promising Results in Phase 1/2 Clinical Trial
YAZIYI PAYLAŞ
Yazar Hakkında
Mehmet Saltuerk
Genetik
Benzer Yazılar
 Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler- Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
- Genetik Hastalıklar: Charcot Marie Tooth (CMT); belirtileri, tipleri ve tedavisi
- Depresyon ve genetik: Mutsuzluk ve intihara neden olan genler
- Kişilik bozukluğu nedir? Türleri, belirtileri ve tedavi seçenekleri
 Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri Genetik Hastalıklar: Charcot Marie Tooth (CMT); belirtileri, tipleri ve tedavisi
Genetik Hastalıklar: Charcot Marie Tooth (CMT); belirtileri, tipleri ve tedavisi Depresyon ve genetik: Mutsuzluk ve intihara neden olan genler
Depresyon ve genetik: Mutsuzluk ve intihara neden olan genler Kişilik bozukluğu nedir? Türleri, belirtileri ve tedavi seçenekleri
Kişilik bozukluğu nedir? Türleri, belirtileri ve tedavi seçenekleri
YORUMUNUZ VAR MI?