
Genetik Hastalıklar: Charcot Marie Tooth (CMT); belirtileri, tipleri ve tedavisi

Charcot-Marie-Tooth, dünyadaki tüm ırklar ve etnik gruplar arasında görülen en yaygın kalıtsal periferik nöropatidir. Charcot-Marie-Tooth veya kısa adıyla CMT, ilk defa 1886 yılıda Jean-Martin Charcot, Pierre Marie ve Howard Tooth tanımlandı. Toplumda görülme sıklığı yaklaşık 1/2500 olan CMT‘nin dünyada 2,8 milyon kişiyi etkilediği tahmin ediliyor.
CMT belirtileri nelerdir?
- İlk işaretler parmaklar ve yürümede: Sık sık ayağa kalkma ihtiyacı, ayak bileği burkulmaları, ayaklarda veya ellerde yanma veya batma hissi.
- Ayaklarda yapısal deformiteler: Ayaklarda yüksek kemerler, içe kıvrık ayak baş parmakları olabileceği gibi bazı hastalarda düz tabanlık da olabilir.
- Ayak dönmesi (foot drop), denge ve yürüme sorunları: Baldır ve ayaklarda kas kaybı ilerledikçe düşme, denge ve yürümede sorunlar olabilir.
Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi
- El becerilerinde zorluklar: Kas kaybı ile ilişkili olarak el becerilerinde zayıflama, örneğin düğme ve fermuar açıp kapamada ve yazı yazmada zorluklar.
- Dokunuşlarda his kaybı: Sıcak ve soğuk arasında ayrım yapamama ve propriosepsiyon kaybı veya vücudun çevre ve uzaysal algı sorunu ve ayrıca birçok insanda nöropatik, kas veya eklem ağrısı.
- Soğuk ve serin veya sıcak ve ılık ayırt etmede zorluklar ve ayrıca çoğu hastada kronik soğuk eller ve ayaklar.
- Ek semptomlar arasında bükülmüş parmaklar ve kontraktürler (elastikiyet kaybı) olabilir: Titreme, diz veya kalça sorunları, kramplar, tenar kası atrofisi (başparmak ile işaret parmağı arasındaki kasların zayıflığı), kas güçsüzlüğü ve el gücünün kaybı, kronik yorgunluk, uyku apnesi, nefes almada zorluklar, yutma güçlükleri, reflekslerin olmaması veya azalması, zayıf dolaşım, skolyoz, kifoz ve işitme kaybı.
- Psikolojik sorunlar: CMT’ye sahip olmanın psikolojik etkisi yıkıcı olabilir. Sinirlilik, depresyon, anksiyete, izolasyon, zevk kaybı, kilo alma, kilo kaybı, umutsuzluk, değersizlik ve suçluluk duyguları olabilir.
CMT teşhisi
Kas fonksiyonu ölçümü, atrofisinin klinik değerlendirmesi, duygusal tepkilerin test edilmesi, elektromiyografik sinir iletim ölçümleri ve de genetik testler ile teşhis edilir.
CMT tedavisi
CMT‘nin şimdilik herhangi bir ilaçlı tedavisi olmamasına rağmen fiziksel terapi, aşırı olmamak kaydıyla orta düzeyde fiziksel aktiviteler kas gücü, dayanıklılığı ve esnekliğini arttırmaya yardımcı olur.
CMT hastaları hangi ilaç tedavilerinden kaçınmalı?
- Amiodarone (Cordarone)
- Arsenic Trioxide (Trisenox)
- Bortezomib (Velcade)
- Brentuximab Vedotin(Adcetris)
- Cetuximab (Erbitux)
- Cisplatin & Oxaliplatin
- Colchicine (extended use)
- Dapsone
- Didanosine (ddI, Videx)
- Dichloroacetate
- Disulfiram (Antabuse)
- Eribulin (Halaven)
- Fluoroquinolones
- Gold salts
- Ipilimumab (Yervoy)
- Ixabepilone (Ixempra)
- Leflunomide (Arava)
- Lenalidomide (Revlimid)
- Metronidazole/Misonidazole (extended use)
- Nitrofurantoin (Macrodantin, Furadantin, Macrobid)
- Nitrous oxide (inhalation abuse or Vitamin B12 deficiency)
- Nivolumab (Opdivo)
- Pembrolizumab (Keytruda)
- Perhexiline (not used in U.S.)
- Pomalidomide (Pomalyst)
- Pyridoxine B6 Vitamininin (Tavsiye edilen günlük miktarın (RDA) 10 katı veya daha fazlası zararlı olabilir. Gıdalarla yüksek miktarda B6 vitamini alımının olumsuz etkilere neden olduğu bildirilmemiştir.)
- Suramin
- Thalidomide
- Zalcitabine (ddC, Hivid)
Depresyon ve genetik: Mutsuzluk ve intihara neden olan genler
Belirsiz veya küçük risk
- 5-Fluoracil
- Adriamycin
- Almitrine (not in U.S.)
- Chloroquine
- Cytarabine (high dose)
- Ethambutol
- Etoposide (VP-16)
- Gemcitabine
- Griseofulvin
- Hexamethylmelamine
- Hydralazine
- Ifosphamide
- Infliximab
- Isoniazid (INH)
- Lansoprazole (Prevacid)
- Mefloquine
- Omeprazole (Prilosec)
- Penicillamine
- Phenytoin (Dilantin)
- Podophyllin resin
- Sertraline (Zoloft)
- Statins
- Tacrolimus (FK506, ProGraf)
- Zimeldine (not in U.S.)
- a-Interferon
Şüpheli risk
- Allopurinol
- Amitriptyline
- Chloramphenicol
- Chlorprothixene
- Cimetidine
- Clioquinil
- Clofibrate
- Cyclosporin A
- Enalapril
- Gluthethimide
- Lithium
- Phenelzine
- Propafenone
- Sulfonamides
- Sulphasalzine
Not: Herhangi bir ilacı almadan veya ilacı değiştirmeden önce, doktorunuzun sizin CMT hastası olduğunuzu bildiğinden emin olmalısınız. Bu ilaçların CMT üzerinde olabilecek olası yan etkilerini konuşmalısınız.
Sinir Sisteminin Organizasyonu
 Sinir Sisteminin Organizasyonu Merkezi Sinir Sistemi (MSS) ve Periferik Sinir Sistemi (PSS)‘den oluşur. Biri olmadan diğeri çalışamaz.
Sinir Sisteminin Organizasyonu Merkezi Sinir Sistemi (MSS) ve Periferik Sinir Sistemi (PSS)‘den oluşur. Biri olmadan diğeri çalışamaz.
MSS, beyni ve omuriliği içerir. Duyusal ve hassas sinyalleri toplar ve işler. Vücudun motor reaksiyonlarını tetikler. PSS ise Sensorik sinirler ile Motor sinirleri içerir. Sensorik sinirler koku, görme, tat gibi duyuları organlardan ve dokulardan beyne taşırken Motor sinirler beyinden iskelet kaslarına ve düz kaslara sinyalleri iletir.
Kısaca özetleyecek olursak;sinyaller Sensorik sinirler aracılığı ile beyne, beyinden gelen sinyaller de Motor sinirler aracılığı ile beyinden kaslara iletilir. CMT hastalığında periferik sinirler olumsuz etkilenir. Bu yüzden genellikle hem motor semptomlara (güçsüzlük ve kas kaybı), hem de duyusal semptomlara (uyuşma) neden olur.
Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
Sinirlerin yapısal hasarları
Periferik sinirlerin akson denilen gövdesi tıpkı bir elektrik kablosu gibi Miyelin kılıf ile sarılarak izole edilmiştir. Eğer Miyelin kılıf hasar görürse sinir uyarıları normalden daha yavaş iletilir. Bu durumdaki hastalar CMT1 olarak tanımlanır. Eğer aksonların kendisi hasar görürse sinir iletim hızı neredeyse normaldir, ancak sinyalin gücü zayıftır. Bu durumdaki hastalar da CMT2 olarak adlandırılır.
CMT Tipleri
CMT tip lerine geçmeden aşağıda bolca geçecek olan teknik bazı terimleri kısaca açıklamak gerekiyor.
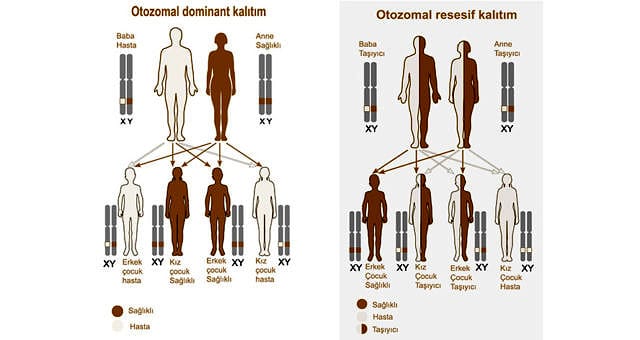
- Otozomal dominant kalıtım: Kromozom çiftleri üzerindeki bulunan aynı genlerden biri mutasyonlu diğeri normal ise bu kişiler heterozigottur ve durum klinik bulgu veriyorsa mutant genler baskındır. Bu tür kalitima Otozomal dominant kalıtım denir.
- Otozomal resesif kalıtım: Hastalığın ortaya çıkması için kromozom çiftleri üzerindeki bulunan aynı genlerin ikisinin de mutasyonlu olmasi gerekiyor. Bu kişiler homozigottur ve bu tür kalitima Otozomal resesif kalıtım denir.
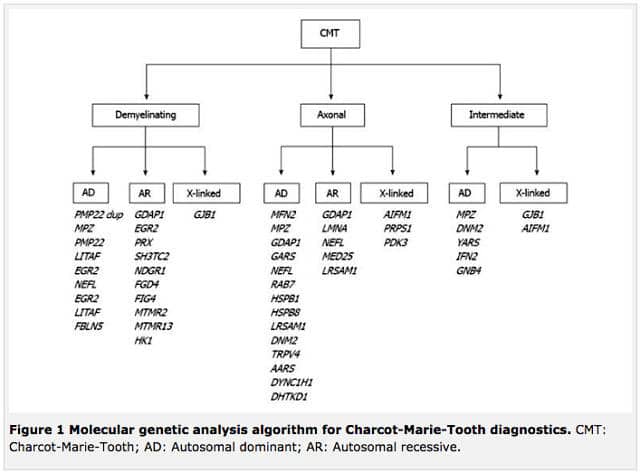
CMT, birçok farklı gende meydana gelen mutasyondan kaynaklanmaktadır. Sebep olduğu gene göre CMT’nin tipi de değişmektedir. 1991 yılından beri yapılan araştırmalarda CMT’ye neden olan 100’den fazla farklı gen tespit edildi ve her geçen gün listeye yeni genler eklenmektedir.
Genetik, klinik, elektrofizyolojik ve histopatolojik kriterlere dayanarak CMT üç ana gruba ayrılır.
- Demiyelinizan form (CMT1)
- Aksonal form (CMT2)
- Distal kalıtsal motor nöropati (dHMN) veya distal olarak da bilinen spinal form Spinal musküler atrofi (dSMA).*
(Spinal musküler atrofi*: Omuriliğin ön boynuzundaki motor sinir hücrelerinin ilerleyen bir şekilde ölmesi.)
Körlük ve kök hücre tedavisi: Yeni geliştirilen yöntem körlüğe çare olabilir
CMT vakalarının çoğunluğu otozomal dominant kalıtımı şeklindedir. Otozomal resesif ve X’e bağlı CMT‘ler daha az yaygındır. Tüm CMT hastalıklarının yaklaşık %50’si PMP22 geninin kopyalanmasından kaynaklanan CMT1A formundan oluşur. Diğer genlerden kaynaklanan CMT formları çok daha az yaygındır.
CMT1
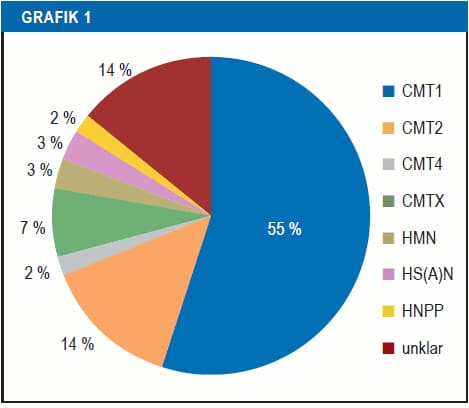 CMT1, sinirleri örten miyelin kılıftaki hasardan kaynaklanır, bu yüzden CMT1 genellikle “demiyelinizan CMT” olarak adlandırılır. CMT1, CMT vakalarının yaklaşık yüzde 55’ini oluşturur. Bu CMT formunun semptomları genellikle çocuklukta başlar ve çoğunlukla ayakları, bacakların alt kısımlarını, elleri ve ön kolları etkiler. CMT 1A hastalarında ilişkili uyku apnesi olabilir. (1) (2). CMT1 vakaları Otozomal dominant *olarak tanımlanır. (Otozomal dominant*: Anne veya babada genin hastalıklı formunun birinin bulunması durumunda çocukların %50 hasta olma ihtimali)
CMT1, sinirleri örten miyelin kılıftaki hasardan kaynaklanır, bu yüzden CMT1 genellikle “demiyelinizan CMT” olarak adlandırılır. CMT1, CMT vakalarının yaklaşık yüzde 55’ini oluşturur. Bu CMT formunun semptomları genellikle çocuklukta başlar ve çoğunlukla ayakları, bacakların alt kısımlarını, elleri ve ön kolları etkiler. CMT 1A hastalarında ilişkili uyku apnesi olabilir. (1) (2). CMT1 vakaları Otozomal dominant *olarak tanımlanır. (Otozomal dominant*: Anne veya babada genin hastalıklı formunun birinin bulunması durumunda çocukların %50 hasta olma ihtimali)
CMT1‘in alt formları
- CMT 1A: 17. kromozomda bulunan PMP22 genindeki bir duplikasyon sorumludur. CMT1’li tüm hastaların yaklaşık yüzde 66‘sını oluşturan en yaygın şeklidir. CMT 1A hastaları genin iki kopyasına sahip olmak yerine (her bir ebeveynden bir tane), kromozomun birinde 2 adet, diğerinde 1 adet kopya bulunur. Bu tipe sahip olanlar çocuklukta yavaş koşuculardır ve yüksek kemerler ile çekiç parmaklar gelişir. Ayak ve bacaklarda problemler ilk 10 yıldan sonra ortaya çıkar. Ayak bileği zayıftır ve değişen derecelerde el zayıflığı vardır. Beynin, uzuvların uzayda nerede olduğunu kestirememesi nedeniyle denge problemleri görülür. Hastaların çoğu (%95‘i) yaşam boyunca normal bir hayat sürer.
Kronik yorgunluk sendromunun bağırsak florası ile bağlantılı olduğu keşfedildi
- CMT 1B: kromozomda bulunan MPZ genindeki bir mutasyon sorumludur. Çocuklarda başlangıçta çok şiddetli semptomlardan, yaşamın ilerleyen zamanlarında ortaya çıkan daha hafif semptomlara kadar geniş bir şiddet aralığı vardır. Bu grupta kalça displazisi ve optik sinir atrofisi ve hastaların yaklaşık üçte birinde skolyoz gözlenmiştir. Erken başlangıçlı CMT 1B’ye sahip hastaların kollarında sinir iletim hızları çok yavaş olup bu hız <15 m/sn‘dir. (Normal> 50 m/sn). Hastalık semptomları bazen 6 ila 20 yaşları arasında başlarken bazen 40 yaşından sonra başlar. Geç başlayan kişilerde hastalık nispeten hafif seyreder.
- CMT 1C: kromozomda bulunan LITAF genindeki bir mutasyon sorumludur. CMT hastalarının %1’den azında görülen nadir bir alt türdür. Semptomlar CMT 1A’ya benzer. İlk belirtiler 1 ile 3 yaşları arasında başlar. Ayaklarda ve ellerde güçsüzlük, atrofi ve duyu kaybı vardır ve sinir iletim hızları yavaştır. (16-25 m/s)
- CMT 1D: kromozomda bulunan ERG2 genindeki bir mutasyon sorumludur. CMT 1D, CMT vakalarının % 1’inden daha azında görülen çok nadir bir alt tiptir. Hastalık bazılarında ciddi semptomlar gösterir. Motor sinir iletim hızları 10 m/s ve daha düşüktür. Bazı CMT 1D vakaları, yaşamın ilerleyen dönemlerinde ortaya çıkar ve daha hafif semptomlar gösterir. Semptomlar arasında Kranial sinir disfonksiyonu* ve solunum zorlukları olabilir. (Bu hastalık bazen Dejerine Sottas sendromu olarak adlandırılır.) (Kranial sinir disfonksiyonu*, beynin alt tarafından ortaya çıkan, kafatasındaki açıklıklardan geçen ve baş, boyun ve gövde kısımlarına yol açan on iki kraniyal sinirden birinin bozukluğudur. Bu bozukluklar, gözler dahil yüzde ağrı, karıncalanma, uyuşma, halsizlik veya felce neden olabilir.)
- CMT 1E: kromozomda bulunan PMP22 genindeki bir noktasal mutasyon sorumludur. CMT 1E de tıpkı CMT 1A gibi PMP22 geninden kaynaklanır ve CMT‘ye sahip kişilerin yaklaşık yüzde birini oluşturur. CMT 1E’li hastalarda semptomlar CMT 1A‘lılara göre daha erken başlar ve daha şiddetli semptomlara sahip olma eğilimindedir, ancak bu değişebilir. Hastalardaki sinir iletim hızları genellikle 10 m/s’nin altındadır ve bu değer önemli ölçüde düşme eğilimindedir. (Kollarda normal ileti hızı > 50 m/s. Çocuklar genellikle iki yaşlarında yürümeye başlar. Pek çok insan erken yaşta yürüteç veya tekerlekli sandalyeye ihtiyaç duyar. CMT 1E, CMT‘nin nadir bir şeklidir ve genetik olarak doğrulanmıştır.
 CMT 1F: kromozomda bulunan NEFL genindeki bir mutasyon sorumludur. CMT 1F, vakaların çok küçük bir yüzdesini oluşturur. Hastalık 8. kromozomun ürettiği Nörofilaman hafif zincir proteinindeki kusurdan kaynaklanan otozomal dominant bir CMT şeklidir.
CMT 1F: kromozomda bulunan NEFL genindeki bir mutasyon sorumludur. CMT 1F, vakaların çok küçük bir yüzdesini oluşturur. Hastalık 8. kromozomun ürettiği Nörofilaman hafif zincir proteinindeki kusurdan kaynaklanan otozomal dominant bir CMT şeklidir.- CMT 1X: X kromozomunda bulunan GJB1 genindeki bir mutasyon sorumludur. Bu alt tip tüm vakaların yüzde 10-16’sını oluşturan ikinci en yaygın CMT şeklidir. Kadınlar, ailelerindeki erkeklerden daha az etkilenirler. Etkilenen kadınların yaklaşık üçte ikisinin hafif aralıkta, üçte birinin orta düzeyde semptomları vardır. Çoğu erkek, yaşlandıkça orta ya da şiddetli nöropatiye sahip olur. CMT 1X’ten etkilenen bir kadının herhangi bir çocuğunun, (çocuğun cinsiyeti ne olursa olsun) hastalığı kalıtım yoluyla çocuğuna verme riski 50/50 dir. CMT 1X olan bir erkek ise hastalığı kızlarının tümüne aktarır. Ancak tüm erkek çocukları sağlıklı olur.
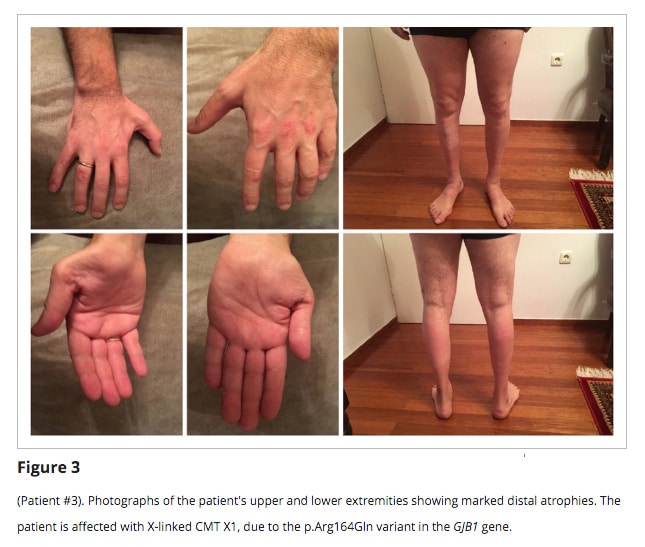
CMT2
CMT2, CMT1’e benzer ancak CMT‘nin daha az yaygın tipidir. CMT2 tipik olarak otozomal dominant bir kalıtım modeli olmasına rağmen bazı durumlarda otozomal resesif bir karekter de gösterebilir (3)(4)(5). CMT2, tüm CMT vakalarının %12 ila %36‘sını temsil eder.(6) CMT2 hastalarındaki semptomlar distal güçsüzlük*, atrofi, duyusal kayıp, azalmış derin tendon refleksleri ve değişken ayak deformitesi ile karakteristiktir (8). CMT2 semptomlarının başlangıcı genellikle CMT1’e göre daha geçtir. Semptomların başlangıcı tipik olarak 5 ila 25 yaşları arasında görülür.(9)(10)
Diyabete karşı keton diyeti! Keton takviyesi ile kan şekeri dengelenir mi?
CMT2, CMT1’in aksine Miyelin kılıfın izole edici aksonlarının hasar görmesinden kaynaklanır. Başka bir ifade ile CMT1 Miyelin kılıfa zarar verirken CMT2 aksonlara zarar verir. Bu yüzden CMT2 “Aksonal CMT” olarak adlandırılır. Distal güçsüzlük*: Gövdenin merkezinden uzakta yer alan alt kol ve bacak, el ve ayak kaslarını kapsayan kaslarda meydana gelen güçsüzlük.
CMT2‘nin alt formları
 CMT 2A: kromozomda bulunan MFN2 genindeki dominant (baskın) bir mutasyondan kaynaklanır. Mutasyona uğramış bu gen hücrede enerji üreten mitokondrilerin füzyonunda olumsuz rol oynar. Açıklama: Dominant formda genin bir kopyası hatalı protein üretirken diğeri kopyası normal protein üretir. Normalde bu durum bazı hastalıklarda avantajdır. Çünkü sağlıklı gen tarafından kodlanan protein birçok hastalıkta kişinin sağlıklı kalması için yeterlidir. CMT 2A’da durum farklıdır, çünkü mutasyonlu protein sağlıklı proteini bloke ederek kişinin CMT hastası olmasına sebep olur.
CMT 2A: kromozomda bulunan MFN2 genindeki dominant (baskın) bir mutasyondan kaynaklanır. Mutasyona uğramış bu gen hücrede enerji üreten mitokondrilerin füzyonunda olumsuz rol oynar. Açıklama: Dominant formda genin bir kopyası hatalı protein üretirken diğeri kopyası normal protein üretir. Normalde bu durum bazı hastalıklarda avantajdır. Çünkü sağlıklı gen tarafından kodlanan protein birçok hastalıkta kişinin sağlıklı kalması için yeterlidir. CMT 2A’da durum farklıdır, çünkü mutasyonlu protein sağlıklı proteini bloke ederek kişinin CMT hastası olmasına sebep olur.- CMT 2A2B: CMT 2A2B de tıpkı CMT 2A gibi MFN2 genindeki mutasyondan kaynaklanır. Ama bu mutasyon başka bir mutasyondur. CMT 2A2B, CMT 2A’nın resesif (çekinik) formudur. Yani CMT 2A2B baskın bir form iken CMT 2A çekiniktir.
- CMT 2B: kromozomda bulunan RAB 7 geni tarafından üretilen RAB 7 proteinindeki bir bozukluktan kaynaklanır. CMT 2B, şiddetli ülserasyon karakterizedir (mide ve onikiparmak bağırsak sorunları). CMT 2B semptomları açık bir şekilde bir duyusal nöropati olmasına rağmen CMT olmadığına dair ciddi bazı görüşler de var.
- CMT 2C: CMT‘nin diğer semptomlarına ek olarak hastalarda diyafram veya vokal kord paralizisi (ses kısıklığı) olabilmektedir. Çok nadir bir formdur. 12. kromozomla bağlantısı bulundu.
- CMT 2D: kromozomda bulunan GARS1 genindeki bir mutasyondan kaynaklanır. CMT 2D kafa karıştırıcı bir bozukluktur, çünkü bazı hastalarda sensorimotor nöropatiler* varken bazılarında sadece motor semptomlar vardır. (Sensorimotor nöropatiler* hem motor, hem de duysal sinir liflerinin etkilendiği nöropatidir.)
- CMT 2E: kromozomda bulunan NEFL genindeki baskın mutasyonlardan kaynaklanır. CMT 2E’yi tedavi etmek için gen düzenleme yöntemi ve akson dejenerasyon yollarını hedefleyen ilaçlar üzerinde çeşitli terapötik yaklaşımlar araştırılıyor.
- CMT 2K: GDAP1 genindeki dominant kalıtsal mutasyonlardan kaynaklanır. GDAP1 geninde mutasyona sahip kişilerin yaklaşık %25’i CMT 2K ve %75’i de CMT 4A‘ya sahiptir ve bunlardaki mutasyon resesifdir. Semptomların başlangıç yaşı değişiklik gösterir. Semptomlar hastaların yaklaşık %25’inde 10 yaşından önce, %41’inde 10-30 yaş arasında, %20’sinde 30 yaşın üzerinde ortaya çıkar. Hastaların sadece %14’ü 50 yaşından sonra semptomsuzdur. CMT 2K hastalarının %10’undan daha azı tam zamanlı tekerlekli sandalyeye ihtiyaç duyar.
- CMT 2O: kromozomda bulunan DYNC1H1 genindeki bir mutasyondan kaynaklanır. Aksonal sinir iletimi çok az veya hiç duyusal semptomu olmayan saf bir motor CMT şeklidir. Şimdiye kadar DYNC1H1 geninde bir tek mutasyon keşfedildi (c.917A> G, p.His306Arg). CMT 2O, spinal musküler atrofi* (SMA-LED) – alt ekstremite zayıflığı, Pes kavus, skolyoz, kalça zayıflığı gibi çeşitli durumlarla ilişkilendirilmiştir. (Spinal musküler atrofi*: Omuriliğin ön boynuzundaki motor sinir hücrelerinin ilerleyen bir şekilde ölmesi)
- CMT 2P: kromozomda bulunan LRSAM1 genindeki mutasyonların neden olduğu nadir bir CMT formudur. CMT‘nin bu türü hakkında literatürde çok fazla yayın yok. Bu nedenle CMT 2P‘ye sahip kişilerin durumunu anlamak için daha fazla araştırmaya ihtiyaç var. Klinik fenotip, yaşamın ikinci veya üçüncü on yılında başlar. Nispeten hafif, çok yavaş ilerleyen bir Aksonal nöropati* olarak özetlenebilir. 30 yaşından sonra çoğu insanda bazı motor semptomlar ve Pes kavus görülür. Ayrıca bacaklarda fasikülasyon veya istemsiz kas seğirmesi ile erkeklerde erektil disfonksiyon rapor edilmiştir. (Aksonal nöropati*: Sinir hücrelerinin gövdesinde yer alan Aksonların zarar görmesidir. Aksonal nöropatili hastalarda ayak ve ayak bileği gibi uzak kaslarda iletim hızı, kollardaki iletim hızından daha az olmasına rağmen nispeten normal sayılır.)
- CMT 2S: kromozomda bulunan IGHMBP2 genindeki bir mutasyondan kaynaklanır. GHMBP2 gen mutasyonları iki farklı fenotip ile ortaya çıkabilir.

- Fenotip: Şiddetli SMARD1 (Solunum Sıkıntısı ile ilişkili Spinal Musküler Atrofi)
- Fenotip: Daha hafif semptonarın olduğu durumdur. CMT 2S resesiftir (çekinik) yani hastalık her iki genin hasarlı olması durumunda ortaya çıkar.
- CMT 2Z: 1 kromozomda bulunan MORC2 genindeki bir mutasyondan kaynaklanır. Bazı vakalar bebeklik döneminde genel halsizlik olarak görürken bazı vakalar yetişkinlikte Proksimal kaslarda* duyu kaybını içerebilen güçsüzlük yaşar. CMT 2Z, CMT‘nin tipik bir örneğidir. Bu form ayrıca kişinin boy uzunluğuna da bağlıdır (yani önce omurgadan en uzaktaki sinirleri etkiler ve daha sonra yukarı doğru hareket eder). Pek çok insanda ilk belirtileri bacaklarda kramplar şeklinde başlar. CMT 2Z’li kişiler, ilerleyen zamanda (beşinci on yılda) tekerlekli sandalyeye ihtiyaç duyar. İlk olarak 2016 yılında CMT ile ilişkilendirildiği için, popülasyonun ne kadarının CMT 2Z’ye sahip olduğunu görmek biraz zaman alacaktır. Ancak çok yaygın bir aksonal CMT formu olabilir. Yayınlanan bir makaleye göre bu gen yüksek mutasyon oranına sahip gibi görünüyor. (Proksimal kaslar*: Vücudun orta hattına en yakın kasları)
CMT3
CMT3, çeşitli kromozomlar üzerinde bulunan MPZ, PMP22, PRX, EGR2 genlerindeki mutasyonlardan kaynaklanmaktadır. Dejerine Sottas Sendromu (DSS) olarak da bilinen tarihsel bir terimdir. Genellikle çekinik ve genellikle belirgin şekilde demiyelinizan (miyelin kaybı) görülür. Sinir iletim hızları <10 m /s‘dir. (Kollarda normal> 50 m/s)
CMT4
Tüm alt tipleri otozomal resesif bir özellik olarak miras alınır. Bazıları hafif, bazıları ağır olmak üzere farklı formları vardır. CMT4’te, gözlerde katarakt ve vücudun bazı bölgelerinde uyuşma gibi de semptomlar görülebilir.
CMT4‘ün alt formları
- CMT 4A: kromozomda bulunan GDAP1 genindeki mutasyonlardan kaynaklanır. Bu gen aynı zamanda CMT‘nin CMT 2K formuna da sebep olmaktadır. CMT 4A formunun semptomları yaşamın ilk yirmi yılında ama çoğunlukla da 10 yaşından önce başlar. Hem alt, hem de üst uzuvlarda kas kaybı ve kontraktürler (elastikiyet kaybı) oluşur. Çoğu hasta 30 yaşına geldiğinde ayak ve bileklerde oluşan hareket kaybı (AFO) nedeni ile baston veya yürüteçe ihtiyaç duyar. Hastaların yaklaşık %75’i 30 yaşından sonra tekerlekli sandalye kullanmak zorunda kalır. Hastalık ilerledikçe, boğuk bir ses gelişebilir. Vokal kord paralizisi bildirilmiştir.
Alzheimer’dan korunma ve alınacak yedi basit önlem! Öneriler ve uyarılar…
- CMT 4B: kromozomda bulunan MTMR2 genindeki bir mutasyondan kaynaklanır. Yapılan sinir biyopsilerinde Miyelin kılıfın fokal olarak katlanmış olduğu tespit edildi. Semptomlar yaşamın erken dönemlerinde (34. ayda) başlar. Çoğu CMT tipinin aksine, hem proksimal, hem de distal zayıflık yaygındır.
- CMT 4C: Kromozomda bulunan SH3TC2 genindeki mutasyonlardan kaynaklanır ve geniş bir şiddet değişkenliğine sahiptir. Duyusal ve motor nöropati çocuklukta başlar.Ayaklar kemerli veya düz taban olabilir. Hastalar genellikle yürümede hafif güçlük çekerler ve bazı hastalar tekerlekli sandalyeye ihtiyaç duyabilir. El ve ayaklarda başlayan güçsüzlük bazen dirsek ve dizlere kadar uzanabilir. İşitme kaybı, ses teli tutulması, yüz felci gibi semptomlar rapor edilmiştir. Kollardaki iletim hızı genellikle 16 ile 36 m/s arasındadır.
- CMT 4D: kromozomda bulunan NDRG1 genindeki bir mutasyondan kaynaklanır. İlk olarak otozomal resesif kalıtıma sahip bir çingene popülasyonunda tanımlandı. Klinik özellikler arasında distal güçsüzlük, kas kaybı ve duyu kaybı, ayak ve el deformiteleri ve derin tendon reflekslerinin kaybı vardı. Bu hastalarda sağırlık genellikle üçüncü on yılda ortaya çıkar. Genç hastalarda sinir iletimi büyük ölçüde azalır.
- CMT 4F: kromozomda bulunan PRX genindeki bir mutasyondan kaynaklanır. İlk olarak büyük bir Lübnanlı ailede tanımlandı. Ciddi bir resesif CMT formudur. Sinir iletimi oldukça yavaştır.
- CMT 4J: kromozomda bulunan FIG4 genindeki bir mutasyonlardan kaynaklanır. İlk olarak 2007 yılında tanımlanan nadir bir form olduğu için hastalığın nasıl ilerlediğine dair elimizdeki bilgiler sınırlıdır. Semptomlar ve başlama yaşı değişkendir. Hastalık, bazı hastalarda klasik bir sunum sergilerken bazılarında, özellikle yetişkinlik döneminde hızlı veya yavaş nöropati sergileyebilir. CMT 4J, otozomal resesif bir formdur, yani semptomların ortaya çıkması için genin her iki kopyasında da mutasyon olması gerekir.
Teşekkürler: Bu makalenin yazım aşamasında benimle deneyim, bilgi ve tecrübelerini paylaşan ve katkı sunan Şakayık Kılıç’a çok teşekkür ederim.
Benzer konularda hazırlanmış diğer makaleler
- Multipl skleroz (MS) nedir? Tanı ve tedavisindeki yeni bilimsel gelişmeler!
- Kırmızı şarap ve siyah üzüm, Multiple Skleroz(MS) hastalarının durumunu kötüleştiriyor.
- Alkolün kas gelişimine olumsuz etkisi
- Multiple Sklerose (MS) ile savaşta vitamin D
- Kokuların anıları çağrıştırması ve koku ile Alzheimer arasında ilişki
- Köpek balığı, Alzheimer ve Parkinson tedavisinde bir umut olabilir
- Alzheimer’a karşı alınacak yedi basit önlem
- Epilepsi hastaları için yeni bir umut : Gen terapisi
- Parkinson tedavisinde kullanılan bir ilaç, Multipl Skleroz tedavisinde de başarılı sonuçlar verdi
Mehmet Saltuerk
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics of the University of Cologne
++++++++++++++++++++++++
Kaynaklar
- Dematteis, M. et al. Charcot-Marie-Tooth disease and sleep apnoea syndrome: A family study. Lancet (2001). doi:10.1016/S0140-6736(00)03614-X
- Dziewas, R. et al. Increased prevalence of obstructive sleep apnoea in patients with Charcot-Marie-Tooth disease: A case control study. Neurol. Neurosurg. Psychiatry (2008). doi:10.1136/jnnp.2007.137679
- Bouhouche, A. et al. A Locus for an Axonal Form of Autosomal Recessive Charcot-Marie-Tooth Disease Maps to Chromosome 1q21.2-q21.3. J. Hum. Genet. (1999). doi:10.1086/302542
- Bouhouche, A. et al. Autosomal recessive axonal Charcot-Marie-Tooth disease (ARCMT2): Phenotype-genotype correlations in 13 Moroccan families. Brain (2007). doi:10.1093/brain/awm014
- Ouvrier, R., Geevasingha, N. & Ryan, M. M. Autosomal-recessive and X-linked forms of hereditary motor and sensory neuropathy in childhood. Muscle and Nerve (2007). doi:10.1002/mus.20776
- Barreto, L. C. L. S. et al. Epidemiologic Study of Charcot-Marie-Tooth Disease: A Systematic Review. Neuroepidemiology(2016). doi:10.1159/000443706
- Reilly, M. M. Axonal Charcot-Marie-Tooth disease: the fog is slowly lifting! Neurology (2005). doi:10.1212/01.wnl.0000173904.97549.94
- Bienfait HME, Verhamme C, Schaik IN, et al. Comparison of CMT1A and CMT2: similarities and differences. J Neurol. 2006;253(12):1572-1580. doi:10.1007/s00415-006-0260-6
- Thomas D Bird M. Charcot-Marie-Tooth Neuropathy Type 2.; 1998. https://www.ncbi.nlm.nih.gov/books/NBK1285/pdf/Bookshelf_NBK1285.pdf. Accessed January 23, 2020.
YAZIYI PAYLAŞ
Yazar Hakkında
Mehmet Saltuerk
Genetik
Benzer Yazılar
 Genetik Hastalıklar: Osteogenezis imperfekta (OI) nedenleri, belirtileri ve tedavisi
Genetik Hastalıklar: Osteogenezis imperfekta (OI) nedenleri, belirtileri ve tedavisi- Genetik Hastalıklar: Ehlers Danlos Sendrom(EDS) nedir? Belirtileri ve tedavisi
- Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
- Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
- Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi
 Genetik Hastalıklar: Osteogenezis imperfekta (OI) nedenleri, belirtileri ve tedavisi
Genetik Hastalıklar: Osteogenezis imperfekta (OI) nedenleri, belirtileri ve tedavisi Genetik Hastalıklar: Ehlers Danlos Sendrom(EDS) nedir? Belirtileri ve tedavisi
Genetik Hastalıklar: Ehlers Danlos Sendrom(EDS) nedir? Belirtileri ve tedavisi Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi
Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi
YORUMUNUZ VAR MI?
Sıkıldım bu hastalıktan ne yapacağımı bilmiyorum