
 Böbrek tümörlerinin %90’ı parankimden (primer böbrek tümörü), %5-10’u ise kaliks ve pelvis renalisten kaynaklanır. Parankimden gelişen primer böbrek tümörleri benign ve malign olarak ikiye ayrılır. Bütün parankim tümörlerinin %80-85’ini renal hücreli kanserler oluşturur.
Böbrek tümörlerinin %90’ı parankimden (primer böbrek tümörü), %5-10’u ise kaliks ve pelvis renalisten kaynaklanır. Parankimden gelişen primer böbrek tümörleri benign ve malign olarak ikiye ayrılır. Bütün parankim tümörlerinin %80-85’ini renal hücreli kanserler oluşturur.
 Böbrek tümörlerinin %90’ı parankimden (primer böbrek tümörü), %5-10’u ise kaliks ve pelvis renalisten kaynaklanır. Parankimden gelişen primer böbrek tümörleri benign ve malign olarak ikiye ayrılır. Bütün parankim tümörlerinin %80-85’ini renal hücreli kanserler oluşturur.
Böbrek tümörlerinin %90’ı parankimden (primer böbrek tümörü), %5-10’u ise kaliks ve pelvis renalisten kaynaklanır. Parankimden gelişen primer böbrek tümörleri benign ve malign olarak ikiye ayrılır. Bütün parankim tümörlerinin %80-85’ini renal hücreli kanserler oluşturur.Erişkinlerde böbrek tümörlerinin klasifikasyonu:
1-Benign tümörler: adenom, onkositom, juksta-glomerüler hücreli tümör, angiomyolipoma, fibroma, leiomyoma, lipom, hemangiom.
2-Malign tümörler:primer tümörler; renal hücreli karsinom, toplayıcı sistem tümörleri, wilms tümörü, sarkomlar (leiomyosarkom, liposarkom, MFH). Sekonder tümörler; adrenal karsinom, retroperitoneal sarkom, pankreas ve kolon kanserleri (direk yayılım), AC, mide, göğüs ve prostat ca (hematojen yayılım), hematolojik tümörler (lenfoma, lösemi), multiple myelom (primer, metastazik).
Sık görülen benign böbrek tümörleri: Kortikal adenom, onkositom, anjiomyolipoma, jukstaglomeruler hücre tümörü.
Renal kortikal adenomlar: Renal tübüler epitelin en sık gözüken tümörüdür. Otopsi çalışmalarında benign böbrek tümörlerinin %7-22’sini oluşturmakta. Lezyonlar genellikle 1 cm’nin altındadır (çoğu 5 mm). Malign-benign epitelyal tümörlerin ayrımına yarayacak kriterler mevcut değil. Papiller histolojik yapıda benign, solid, parankimal lezyonlardır. Bütün solid renal epitel kaynaklı tümörler potansiyel olarak malign olarak düşünülmeli ve bu şekilde tedavi edilmelidir. Cerrahi olarak renal eksplorasyon ve wedge rezeksiyon uygulanır.
Renal onkositomlar: Solid renal tümörlerin %3-7’sini oluşturur. Erkeklerde 2 kat fazladır. Genetik olarak kromozom 1 ve Y kaybı, 11q13’de translokasyon vardır. Renal hücreli kanserlerin aksine 3’üncü kromozom kaynaklı herhangi bir genetik anomali söz konusu değildir. Boyut olarak genellikle 4-6 cm arasında değişir. Makroskopik olarak santral skar formasyonu genellikle bulunur. Açık kahve renkli ve kapsüllüdür. Histopatolojik olarak geniş eozinofilik sitoplazma içinde bol mitokondri ile karakterizedir (onkosit). Distal renal tübüllerden kaynaklanır. Ayırıcı tanıda özellikle kromofob hücreli renal kanserler önemli. Multisentrik ve bilateral olabilirler (%4-13).
Tanı: BT; santral satellite skar görünümü vardır. Anjiografi; tekerlek manzarası görünümü tanıda yardımcıdır. Preoperatif tanı koymak neredeyse imkansız.
Tedavi: renal eksplorasyon, radikal veya parsiyel nefrektomi.
Anjiomyolipoma (hamartom): Matür yağ dokusu, düz kas hücreleri ve kalın duvarlı damarlardan meydana gelen benign tümörlerdir. Orta yaşlarda ve kadınlarda (4 kat) daha sık gözükür. Kapsüllü, sarı-gri renktedir. Gelişiminde hormonal etkinin rolü olabileceği söyleniyor ancak kesin değil.
Tuberoskleroz sendromu: mental retardasyon, epilepsi, beyinde gliozis, yüzde adenoma sebaseum, submukozal fibrozis, retina, AC, KC, pankreas, kemikte anjiomyolipomlar ile seyreden otozomal dominant hastalık. ———–Anjiomyolipomların %20’si tuberosklerozlu hastalarda görülür ve tuberosklerozlu hastaların %50’sinde anjiomyolipom vardır. Bu özel grupta hastalar daha erken yaşlarda (30 yaş civarı) tanı alırlar ve tümörler bilateral ve multisentrik olmaya eğimlidir.
Semptomlar: yan ağrısı, hematüri, palpabl kitle, hipovolemik şok. Hastaların %10’unda masif retroperitoneal kanama görülebilir (Wunderlich sendromu).
Tanı: ultrasonografi ve özellikle bilgisayarlı tomografi faydalı.
Tedavi: Asemptomatik, 4 cm altı tümörlerde; takip ve 6-12 aylık aralıklarla kontrol yapılır. Daha büyük ve semptomatik tümörlerde; eksplorasyon (parsiyel nefrektomi veya embolizasyon tercih edilmeli).
Jukstaglomeruler tümör: Çok nadirdir. Hipertansiyona neden olur. Genç yaşta ortaya çıkar, tömür jukstaglomeruler bölgedeki perisitlerden köken alır. Bu hücreler özel boyalar ile boyandığında renin granülleri içerdiği görülür. Parsiyel nefrektomi ile tedavi edilir.
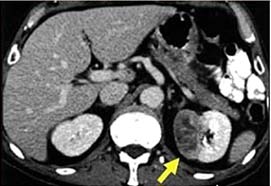
Renal hücreli karsinom
Epidemiyoloji: Bütün erişkin tümörlerinin %3’ünü oluşturur. Böbreğin malign tümörlerinin %85’ini oluşturur. Üriner sistem kaynaklı maligniteler arasında 3. sıklıkta gözükür. Erkek/kadın oranı: 3/2. Daha çok 60-70 yaşları arasında görülür. Ortalama 5 yıllık sağ kalım %60 civarındadır. Son yıllarda Amerika ve Avrupa’da görüntüleme yöntemlerinin yaygın olarak kullanılması nedeniyle insidental böbrek tümörü tanısı alan hasta sayısı artmakta. İnsidental olarak yakalanan böbrek tümörleri genellikle boyut olarak daha küçük ve erken evrede. Ülkemizde bu konudaki veriler net değildir.
Etyoloji: Sigara kullanımı: risk %30-50 artmakta, risk kullanılan miktarla doğru orantılı olarak artmakta.
Obezite: belirgin ilişki sadece ileri derecede obez olan kişilerde gösterilmiş (BMI >4. kadranda). Endojen östrojen miktarının ve serbest insülin-like growth faktörün artması. Son dönem böbrek yetmezliği (KBY), acquired renal kistik hastalık (ARCD).——-Risk faktörleri; hipertansiyon, ilaç (tiazid cinsi diüretiklerin kullanımı, analjezikler), mesleksel (demir, çelik, kadmium, petrol, asbestos sanayinde çalışanlar), diyet (kahve, kızarmış, kömürde pişmiş etler), hormonlar (dietilstilbesterol kullanımı), enfeksiyonlar, polikistik böbrek hastalıkları.
Genetik: Renal hücreli karsinomun (RHK) sporadik ve ailesel (herediter) olmak üzere iki şekli vardır. Major genetik anomali kromozom 3p kaybıdır. Hem herediter,hem sporadik tipte sıktır. Papiller RHK; 7. ve 17. kromozom anomalisi vardır. P53, Rb1 geni bozuklukları önemlidir. Wilms tümör geni (WT1) ve NM23 tm baskılayıcı gen anormalileri RHK’da görülür. VHL tümör baskılayıcı geni anormalileri önemlidir.
Von Hippel Lindau (VHL) sendromu: Böbrek hücreli kanserin, şeffaf hücreli biçiminin ailesel formunu oluşturur. Otozomal dominant bir hastalıktır. Görülme sıklığı 1:36000. Bilateral ve multifokal olma oranları yüksektir.
Von Hippel-Lindau sendromunda bulunabilenler: Retinal anjiyom, hemanjiyoblastom, böbrek hücreli kanser, böbrek kistleri, feokromasitoma, adenokanser, pankreas kisti, kistadenom, endolenfatik kese tümörü.
Patoloji: Renal hücreli karsinom tarihsel olarak Grawitz tümörü, hipernefroma, renal adenokarsinom olarak ta adlandırılmıştır. 1997 Heidelberg klasifikasyonuna göre patolojik olarak 5 alt gruba ayrılmaktadır. Çoğu proksimal tubül kaynaklı, kromofob ve toplayıcı kanal kanserleri ise distal tubül kaynaklıdır. Tipleri; konvansiyonel RHK (şeffaf ve granüler hücreli dahil), papiller RHK (kromofilik), kromofob RHK, toplayıcı kanal RHK (medüller kanser dahil), sınıflandırılamayan.
Konvansiyonel renal hücreli kanser: en sık gözüken tip (%70). Kökeni proksimal kıvrıntılı tüpler. %4 multifokal, %0.5-3 bilateral olabilir. Sitoplazmalarında bol miktarda lipid ve glikojen ihtiva etmeleri nedeniyle tipik olarak sarı renkli. Nükleus küçük ve koyu renkli.
Papiller renal hücreli kanser: ikinci en sık gözüken tip (%10-15). Kökeni distal kıvrıntılı tüpler. Sitogenetik anomaliler sık. Multifokalite oranı yüksek (%39). Tip 1 ve 2 olmak üzere iki cinsi vardır.
Kromofob renal hücreli kanser: üçüncü sıklıkta (%5). Kökeni kortikal toplayıcı kanallar. Prognozu çok iyi. Genellikle unilateral. Ayırıcı tanıda onkositom önemli.
Toplayıcı kanal renal hücre kanseri: en az sıklıkta gözüken (%1). Kökeni medüller toplayıcı kanal. Çok agresif seyreder, prognozu çok kötü. Genellikle çok büyük hacimlere ulaşmaz.
Semptom ve belirtiler: Klasik triad; hematüri (%40-60), yan ağrısı (%40), palpabl kitle (%20-30).
Renal hücreli kanserde paraneoplastik sendromlar (%40): hiperkalsemi, polisitemi, hipertansiyon, stauffer sendromu, kaşeksi, kilo kaybı, ateş, amiloidoz, prolaktin ve glukagon hormonlarında artış.
RHK’da sistemik bulgular: sedim artışı, hipertansiyon, anemi, kaşeksi, kilo kaybı, anormal karaciğer fonksiyonu, hiperkalsemi, polisitemi, nöromiyopati, amiloidoz.
Fizik muayene: Palpabl kitle, Varikosel, Supraklavikular/servikal adenopati, Alt ekstremite ödemi.
Renal hücreli kanserde hangi hastalarda tarama yapılmalı: Son dönem böbrek yetmezliği olan hastalar, VHL sendromu olan hastalar, VHL sendromu ve ailesel RHK olan hastaların akrabaları, tüberosklerozlu hastalar.
Renal hücreli kanserde TNM sınıflaması:
T1: Tümör böbrekte sınırlı, en büyük hacmi <7 cm.
T1a: Tümör böbrekte sınırlı, en büyük hacmi <4 cm.
T1b: Tümör böbrekte sınırlı, hacmi 4-7 cm arası.
T2: Tümör böbrekte sınırlı, en büyük hacmi >7 cm.
T3: Tümör major venlere, adrenal beze ve perinefritik yağ dokusuna yayılım gösterir ancak gerota fasyasını geçmemiştir.
T3a: Adrenal beze ve perinefritik yağ dokusuna yayılım var, gerota fasyası geçilmemiştir.
T3b: Renal ven veya diyafragma altı vena cava tutulumu mevcut.
T3c: Diyafragma üstü vena cava tutulumu mevcut.
T4: Tümör gerota fasyasını geçmiştir.
N: regional lenf nodları (hilar, paraaortal, paracaval).
N0: Lenf nodu metastazı yok.
N1: Tek bir lenf nodu metastazı var.
N2: Birden fazla lenf nodunda metastaz var.
M: Uzak metastaz
M0:Uzak metastaz yok.
M1:Uzak metastaz var.
TNM ve Robson sınıflaması:
Stage 1: T1-2; böbrekte sınırlı tümör.
Stage 2: T3a; gerotayı geçmemiş, adrenal beze ve perinefritik yağ dokusuna yayılım.
Stage 3a: T3b-c; gerotayı geçmemiş majör venlere yayılım.
Stage 3b: N1-2; nodül tutulumu.
Stage 4a: T4; gerotayı aşmış tümör.
Stage 4b: M1; uzak metastaz.
Böbrek tümörlerinde metastaz: Akciğer %50-60. Kemik %30-40. Bölgesel lenf nodu %15-30. Renal ven %10-20. Perirenal yağ doku %10-20. V.cava %8-15. Komşu organ %20. Karşı böbrek %10.
Tanı:
1-Semptom ve belirtiler.
2-Laboratuar bulguları: ideal marker yoktur. Anemi, eritrosit sedimantasyon hızında artış olur. Ferritin, NSE (nöron spesifik enolaz), GAG, CEA, CA19-9, CA15-3 artar. GGT; metastazik hastalıkta yükselir. ALP; kemik metastazında yükselir.
Görüntüleme yöntemleri:
İntravenöz pyelografi (IVP): yalnız başına kullanıldığında sadece %65-70 sensitivitesi vardır. Sensitivite özellikle <3 cm tümörlerde çok düşük. Tipik görüntü; kitle etkisi. Tanıyı kanıtlamak için ilave radyolojik tetkiklere ihtiyaç vardır. IVP’de normal bulgular olması renal kitle varlığını ekarte etmez.
Ultrasonografi (USG): non-invaziv bir yöntem. Renal hücreli kanser için spesifik bir USG bulgusu yok. Sensitivitesi %80 civarında. Ana kısıtlama ufak tümörlerde (<1.5 cm). Özellikle evrelemede yetersiz. Böbrek kistlerini tanımlamada %98 doğruluk oranı vardır.
Bilgisayarlı tomografi (BT): böbrek tümörlerin tanısında ve evrelemesinde altın standart, doğruluk oranı diğer yöntemlere göre çok yüksek. Sensitivite ve spesifitesi %95’ler civarında. Ufak tümörlerin tanısında da aynı derecede doğruluk oranlarına sahip.
Magnetik rezonans inceleme (MRI): böbrek tümörlerinde vasküler yayılımın belirlenmesinde en etkili radyolojik görüntüleme yöntemi. İntrarenal (T2) ve ekstrarenal (T3a) tümör ayrımında BT’den üstün. Multiplanar kesitler alınması nedeniyle tümörün tam olarak lokalizasyonun belirleme avantajı nedeniyle özellikle parsiyel nefrektomi yapılacak hastalarda kullanılmakta.
Metastaz bulguları varlığında şunlar yapılır; karaciğer fonksiyon testleri, akciğer tomografisi, (akciğer grafisine ek olarak), beyin tomografisi, kemik sintigrafisi.
Ayırıcı tanı: Renal adenom, onkositom, angiomyolipoma, abse, infarkt, damar malformasyonları, psödotümör, metastaz.
Lokalize böbrek tümörlerinde tedavi: RHK’de primer tedavi cerrahidir. Kemoterapi ve radyoterapiye dirençlidirler. Cerrahi iki şekilde uygulanabilir.
1-Radikal nefrektomi: açık veya laparoskopik.
2-Parsiyel nefrektomi (nefron koruyucu cerrahi): Radikal nefrektomi; böbreğin ipsilateral adrenal bez, perirenal yağ dokusu, gerota fasyası, bölgesel lenf nodları ve üreter 2/3 üst bölüm ile birlikte çıkarılmasıdır. Transperitoneal, torakoabdominal veya subkostal kesi ile yapılabilir.
Nefron koruyucu cerrahi hangi hastalarda uygulanabilir:
Kesin endikasyonlar: soliter böbrekte tümör varlığı, bilateral RHK, familyal RHK’de multifokal tümör varlığı (VHL).
Relatif endikasyonlar: karşı böbrekte renal arter darlığı, hiddonefroz, tekrarlayan pyelonefrit atakları, taş hastalığı, nefroskleroz bulunması.
Elektif endikasyonlar: karşı böbreği normal olan hastada <4 cm tümör bulunması.
Metastatik böbrek tümörlerinde tedavi: Hastaların %25’i ilk tanı anında metastatik böbrek tümörü tanısı alır, %25’inde ise tedaviden sonra metastatik hastalık gelişir. Beklenen ortalama yaşam süresi çeşitli faktörlere bağlı olmak üzere 6-24 ay arasında değişir; 6 ay – %73, 12 ay – %48, 60 ay – %5-8.
Standart tedavi: Cerrahi (sitoredüktif nefrektomi) + Sistemik terapi (immunoterapi; INF-alfa, İL-2, retinoidler).
Metastatik böbrek tümörlerinde tedavi (devam):
Sitoredüktif nefrektomi: yapılan çalışmalarda ortalama 10 aylık bir survival avantajı sağlamakta.
Metastazektomi: özellikle soliter akciğer metastazı olan hastalarda sonuçlar daha iyi.
İmmunoterapi: interferonlar, İnterlökin-2, tümör infiltre edici lenfositler, kombine tedaviler
Radyoterapi: metastazların tdv’sinde hastaların 2/3 ‘ünde etkili (palyatif tedavi).
Gen tedavileri: henüz çalışma aşamasında.
Prognoz: 5 yıllık sağ kalım oranları; Evre I: %90-100. Evre II: %75-90. Evre III: %60-70. Evre IV: %15-30.
Wilm’s tümörü (nefroblastom):
Wilms tümörü (WT) metanefrik blastemin anormal proliferasyonu sonucu meydana gelir. Çocuklarda üriner traktın en sık görülen malign tümörüdür. Solid tmlerin %8’i, üriner sistem tmlerinin %80’ini oluşturur. En sık 3-4 yaş. Kadın = erkek. Yıllk insidans 15 yaş altı çocuklarda 1.000.000’da 7-10’dur.
Etyoloji: Embriyonel hayatta metanefrik blastemin anormal gelişimi sözkonusudur. 11. kromozom kısa kolunda delesyon olabilir. Normal renal gelişimde rol oynayan WT1 (11p13) ve WT2 (11p15) genlerindeki hasar varlığı Wilms tümörü olşumunda rol oynar. Trizomi 18 ve 8 olabilir. Wilms tümörlü hastaların yaklaşık %10-15’i doğumsal anomalilere sahiptir. Bunlar aniridi, hemihipertrofi veya hızlı büyüme sendromları ve genitoüriner anomalilerdir. Kas iskelet sistemi yüz ve kafatası anomallileride olabilmektedir. Wilms tümörü ile ikincil tm olarak AC, kolon, serviks ve başka organ kanserleride görülmektedir.
Wilms tümörü ile birlikte görülen sendromlar:
Denys-Drash sendromu: Wilms tümörü, genital ambiguite (ambigius genitalia), progressif glomerulopati.
Beckwith-Wiedemann sendromu: visseromegali, hemihipertrofi, omfalosel, mikrosefali ve makroglossi.
WAGR: Wilms tümörü, aniridi, genitoüriner anomaliler, mental retardasyon.
Satos sendromu: makrosefali ve wilms tümörü.———-WT1 geninin kaybı Denys-Drash sendromu ve WAGR sendromu ile ilişkili iken; Beckwith-Wiedemann sendromunda WT2 gen kaybı vardır.
Patoloji: Enkapsüler, soliter bir böbrek tümörüdür. Tümörün yüzeye doğru çıkıntı yaptığı gözlenir. Kesitlerde hemoraji, nekroz alanları ve kistik formasyon izlenebilir. Mikroskopi; metanefrik ve mezodermal hücrelerden oluşur. En önemli özelliği stromal hücreler içinde helezonvari nefrojenik hücrelerdir. Stromada düz kas, kıkırdak ,yağ ve kemik hücreleri görülebilir.
Sınıflama:
A-Atipik formlar: 1-Anaplazik tip (%2). 2-Rabdoid tip (%2); mortalitesi çok yüksektir, beyin ve sinir sistemine metastaz yapar. 3-Clear cell sarkomu (%3); kemiklere metastaz yapar.
B-Tipik formlar: 1-Multiloküler kist formu; yetişkinlerde özellikle kadınlarda sık görülür, benign seyirlidir. 2-Konjenital mezoblastik nefroma; benign seyirlidir, leimyoma benzer. 3-Rabdomyosarkom tipi.
Klinik ve tanı: Hasta çoğunlukla tesadüfen farkedilen kitle veya karın şişliği ile getirilir. Karın ağrısı, gözle görülür hematüri, kitle rüptürüne bağlı akut batın, yüksek ateş, HT, kabızlık veya kilo kaybı eşlik eden diğer bulgulardır. Tanı; USG ve İVP yeterlidir. Ancak böbreğin görülemediği durumlarda BT ve ranal anjiografi yapılabilir.
Evreleme (NWTS):
Evre 1: tümör böbreğe sınırlı, renal kapsül intakt, tümör operasyon esnasında parçalanmamıştır, rezidü tümör yoktur.
Evre 2: tümör böbreği aşmıştır, ancak tamamen çıkarılmıştır. Böbrek damarlarında, çevre dokularda invazyon vardır, ancak tamamen çıkarılmıştır.
Evre 3: batına nonhematojen yayılım söz konusu. A)Böbrek hilusu, paraaortikler ve ötesinde biyopside tutulum var. B)Operasyon öncesi veya esnasında peritona diffuzyon veya kontaminasyon var. C)Periton yüzeyinde implantasyon var. D)Tümör mikroskobik veya gross olarak çıkarılamayacak kadar yayılmış. E)Tümör vital sahaları invaze ettiği için çıkarılamamıştır.
Evre 4: evre 3’e ek olarak hematojen yayılım var (AC, KC, kemik, beyin, vs).
Evre 5: tanı anında bilateral böbrek tutulumu var.
Prognoz: Küçük yaşlarda tedaviye daha iyi cevap verir. 2 yaş altı prognoz daha iyidir. Nüks yada metastazsız iki yıllık sağkalım tam şifa olarak kabul edilir.
Tedavi: Evre 1-2 tipik formlar ve evre 1 anaplazik tip: cerrahi + kemoterapi (actinomycin D + vincristin). Evre 3-4 tipik formlar ve clear cell sarkomun tüm evreleri: cerrahi + 1080 Gy radyoterapi + kemoterapi (actinomycin D + vincristin + doxurobicin). Evre 2-3 anaplazik tip: cerrahi + radyoterapi + kemoterapi (3’lü), radyoterapiye cerrahiden sonraki 1-3 gün içinde başlanır, metastazlara da RT uygulanır. Atipik formlarda 2000 cGy, evre 3 tipik formlarda 1000 cGy, evre 2-3-4 anaplazik tip 1200-4000 cGy verilir.
YAZIYI PAYLAŞ
Yazar Hakkında
Dr. Enes Başak
Aile Hekimliği
Benzer Yazılar
 Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri- Böbrek kanseri neden olur? Belirtileri ve en etkili tedavi yöntemleri
- Renal hücreli kanser tedavisinde yeni terapilerle neler değişti?
- Metastaz nedir, neden olur? Metastatik kanser belirtileri ve tedavisi
- Kanseri ilacı Afinitor Türkiye’de de ruhsat aldı
 Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri Böbrek kanseri neden olur? Belirtileri ve en etkili tedavi yöntemleri
Böbrek kanseri neden olur? Belirtileri ve en etkili tedavi yöntemleri Renal hücreli kanser tedavisinde yeni terapilerle neler değişti?
Renal hücreli kanser tedavisinde yeni terapilerle neler değişti? Metastaz nedir, neden olur? Metastatik kanser belirtileri ve tedavisi
Metastaz nedir, neden olur? Metastatik kanser belirtileri ve tedavisi Kanseri ilacı Afinitor Türkiye’de de ruhsat aldı
Kanseri ilacı Afinitor Türkiye’de de ruhsat aldı
YORUMUNUZ VAR MI?