
Genetik hastalıklar: Dravet sendromu nedir? Nedenleri ve tedavisi

Dravet sendromu, bebeklik veya erken çocukluk döneminde başlayan hafiften şiddetliye kadar değişen semptom yelpazesi gösteren ve bireyleri yaşamları boyunca etkileyen bir epilepsi çeşididir. Toplumda görülme sıklığı yaklaşık 1:15.700 kişidir. Nöbetler genellikle bir yaşından önce başlar. İlk nöbetler genellikle uzun sürelidir ve vücudun yarısını etkiler. Daha sonraki nöbetler vücudun diğer tarafına geçebilir. Çocuklar yaşamlarının ilk birkaç yılında normal gelişim göstermelerine rağmen nöbetler arttıkça ve yaş ilerledikçe gelişimde yaşıtlarının gerisinde kalmaya başlar. Yaşamın ikinci veya üçüncü yılına kadar gelişimsel gecikme ve anormal EEG ler gibi diğer komorbiditeler genellikle belirgin değildir.
Dravet sendromu 1989’dan önce Epilepsy with polymorphic seizures (polimorfik nöbetli epilepsi), Polymorphic epilepsy in infancy (PMEI) (bebeklik dönemi polimorfik epilepsi) veya Severe myoclonic epilepsy in infancy (SMEI) (bebeklik dönemi şiddetli miyoklonik epilepsi) olarak biliniyordu.

Yaygın görülen sağlık sorunlar
- Uzun ve sık nöbetler.
- Davranışsal ve gelişimsel gecikmeler.
- Hareket ve denge sorunları.
- Ortopedik sorunlar.
- Gecikmiş dil ve konuşma sorunları.
- Büyüme ve beslenme sorunları.
- Uyku güçlükleri.
- Kronik enfeksiyonlar.
- Duyu bütünleme bozuklukları.
- Vücut ısısı, kalp atış hızı, kan basıncı ve diğer sorunları düzenlemede güçlüklere yol açabilen disotonomi veya otonom sinir sistemi bozukluklar.
Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
İkincil sağlık sorunları
Bazı durumlarda hastadan hastaya değişen şiddette ikincil sağlık sorunları görülebilir.
- Kardiyovasküler sorunlar
- Diş sağlığı sorunları
- Disotonomi (İstemli sinir sisteminde işlev yetersizliği)
- Ortopedik ve skolyoz sorunları
- Uyku bozuklukları
- Zayıflamış bağışıklık
2015 yılında yapılan bir çalışmaya göre aşağıdaki kriterlerden en az 4’ünü gösteren hastalara Dravet sendromu tanısı konuyor.
- Nöbetlerin başlangıcından önce normal veya normale yakın bilişsel ve motor gelişim
- 1 yaşından önce ateşli veya ateşsiz iki veya daha fazla nöbet.
- Miyoklonik, hemiklonik veya jeneralize tonik-klonik nöbetlerden oluşan nöbet öyküsü
- 10 dakikadan uzun süren iki veya daha fazla nöbet.
- 2 yaşından sonra nöbetlerin devam etmesi durumunda birinci basamak antiepileptik ilaç tedavisine yanıt vermeme.
Sendromun diğer belirtileri arasında aşılarla karşı gelişen nöbetler, sıcak banyo ve sıcak havayla ilgili problemler, gelişimsel yavaşlama veya durgunluk veya yaşamın ilk yılından sonra gelişimde gerileme, davranışsal sorunlar ve konuşmada gecikme.
Kromozomal hastalıklar ve gebelik: 35 yaş üstü hamilelikler neden riskli
Dravet sendromlu bebekler başlangıçta normal gelişim gösterirler ve yukarıdaki kriterlerin birçoğu yaşamın ilk yılında belirgin değildir, bu yüzden aşağıdaki semptomlardan birini gösteren bebekler genetik test yapılması düşünülmelidir:
- 1 yaşına kadar 2 veya daha fazla uzun süreli nöbet.
- 1 yaşına kadar 1 uzun süreli nöbet ve herhangi bir hemiklonik nöbet (vücudun bir tarafında sürekli, ritmik sarsıntı).
- Vücudun çeşitli yerlerinde etkileyen kısa veya uzun 2 nöbet.
- 18 aylıktan önce nöbet öyküsü. Kollar veya bacaklarda hızlı ve istem dışı uzun veya kısa süreli kasılma nöbetleri (Miyoklonik, absens nöbetler).
Eğer çocuğunuz veya bir yakınınızda bu belirtiler varsa Dravet sendromundan şüphelenilmeli ve bir nöroloji uzmanı görüşülmelidir.
Sebep: Genetik
Birkaç gende meydana gelen mutasyonun Dravet sendromuyla ilişkili olduğunu biliniyor. Bu genlerden SCN1A, SCN2A ve SCN9A genleri hücre zarında bulunan ve iyon giriş çıkışından sorumlu olan sodyum kanalları (Na)ilgili genlerdir. GABRG2 geni ise GABA reseptörü ile ilişkili gendir.
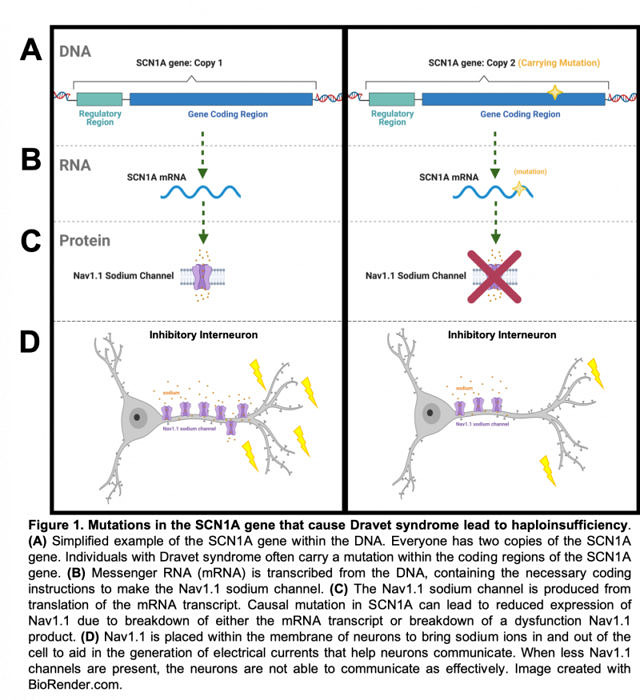
Dravet sendromu vakalarının %90’ından fazlasında SCN1A geninde meydana gelen anormal dizilimden kaynaklanır. Bu anormal dizilim, bazen bir mutasyon (bir veya daha fazla nükleotidin başka nükleotidler ile yer değiştirmesi), bazen bir delesyon (bir veya daha fazla nükleotidin kaybolması), bazen de duplikasyon (bir veya daha fazla nükleotidin tekrarı şeklinde olur. Her üç şekilde de kodlanan Nav1.1 proteini hatalıdır.
Genetik Hastalıklar: Charcot Marie Tooth (CMT); belirtileri, tipleri ve tedavisi
Hastalığın şiddeti bu değişikliklerin yani mutasyon, delesyon veya duplikasyonun) nerede olduğuna bağlı olarak değişir. Dravet sendromu nadiren de olsa SCN1B, GABRG2 veya HCN1 gibi diğer genetik mutasyonlarla ilişkili olabilir. Klinik görünüm benzer olsa da bazı hastalar alternatif tedavi yolları gerektirebilir.
Bazen genetik testlerde bir mutasyon, delesyon veya duplikasyon bulunmayabilir ancak doktorlar semptomlara dayanarak Dravet sendromu teşhisi koyabilir.
Mutasyonların kaynağı ve etkileri:
- SCN1A mutasyonlarının %90’ı de novo’dur, yani hastanın ebeveynlerinde bulunmazlar.
- SCN1A mutasyonlarının %4-10’u ebeveynden alınır. Bu durumda mutasyonun çocuklara geçme olasılığı %50’dir.
- SCN1A geni 6.000 civarında nükleotidten oluşur, dolayısı ile bir mutasyonun gerçekleşebileceği çok fazla yer vardır. Bu nedenle, çoğu hastanın mutasyon noktası başka bir hasta ile aynı değildir, bu da hastalığın şiddetinin hastadan hastaya farklılık göstermesine sebep olur.
- SCN1A geninde görülen her mutasyon Dravet sendromuna sebep olmaz. Daha açık bir ifade ile bu tür mutasyonlar amino asit diziliminde bir değişikliğe sebep olmayan zararsız mutasyonlardır ve protein yapısını değiştirmezler.
- SCN1A mutasyonları ayrıca migren, ateşli nöbet(FS) ve ateşli nöbet+epilepsi (GEFS+) ile ilişkilidir.
SCN1A geni ve Dravet sendromuna etkisi
SCN1A geninin görevi nedir: SCN1A geni hücre zarında yer alan ve sinir sisteminin düzenli işleyişinde rol oynayan sodyum kanallarını kodlayan bir gen dir. Her sağlıklı kişinin beyninde bir işlem gerçekleşeceğinde bu kanallardan (+) ve (-) yüklü iyonlar hücre içine ve dışına belirli bir düzen çerçevesinde girer ve çıkarlar.
Depresyon ve genetik: Mutsuzluk ve intihara neden olan genler
Sinirler arası bilgi akışı ve iyon kanallarının rolü
Beynimizde sinirler arası iletişim sinir hücreleri arasında elektrik akımı kullanılarak gerçekleştirilir. Bu elektrik akımları, sodyum, potasyum, kalsiyum ve klorür gibi pozitif (+) ve negatif (-) yüklü iyonlar aracılığı ile sürdürülür.
Sinir hücreleri, elektrik taşıyan kablolara benzerler ve birbirleri ile iletişimde + yüklü Sodyum (Na+) ile + yüklü potasyum (K+) önemli rol oynar. Sinir hücresi komşu sinir hücresi ile bir iletişimde bulunmazken içi negatif yüklüdür. Sodyum (Na+) ve potasyum (K+) gibi pozitif yüklü iyonlar ise çoğunlukla hücrenin dışında tutulur.
Bir sinir hücresi uyarıldığında yani bir mesaj göndermesi gerektiğinde aniden bir elektrik akımı başlar ve bunun için sodyumun (Na+) hücreye girmesine izin verir. Pozitif ve negatif iyon dengesindeki bu değişiklikler elektrik akımına yol açar ve bu da komşu sinir hücre ile iletişim kurulmasını sağlar. İletişimi kesmenin zamanı geldiğinde bu süreç tersine çevrilir. Böylece içeride daha fazla negatif, dışarıda daha fazla pozitif yüklü iyonlar yer alır. İyonların hücre içinde ve dışında dengelenmesi hücrenin elektriksel aktiviteyi durdurması için önemlidir ki, bu daha fazla pozitif yüklü iyonun dışarıya taşınması anlamına gelir.
İyonların hücre içine ve dışında hareket etmeleri için hücrelerin özel kanallara ihtiyacı vardır. Bu kanallar Sodyum (+Na) için ayrı, potasyum (K+) dir. Yani her iyon için özelleşmiş iyon kanalına ihtiyaç vardır. Bu kanalların düzgün çalışmaması veya yeterli sayıda olmaması durumunda sinir hücreleri elektrik akımlarını düzenleme, iletme ve durdurmada zorlanır. Bu düzensiz elektrik akımları ise nöbetlerin ortaya çıkmasına yol açar.
Genetik Hastalıklar: Ehlers Danlos Sendrom(EDS) nedir? Belirtileri ve tedavisi
Sodyum kanalının bozulması nöbetlerin ortaya çıkmasına sebep oluyor
Dravet sendromunun en yaygın nedeni SCN1A genin kodladığı Nav1.1 adındaki sodyum kanalında meydana gelen bir problemden kaynaklanır. Birçok Dravet sendromlu hastada, Nav1.1’i kodlayan iki SCN1A geninden en az birinde mutasyon bulunur. SCN1A geninin bir kopyayında meydana gelen mutasyon Nav1.1 sodyum (Na +) kanallarının kabaca yarısının düzgün çalışmamasına veya hiç bulunmamasına sebep olur. Bu durum pozitif ve negatif yüklü iyonların hücreye düzenli bir şekilde girip çıkmasında problemlere neden olur ki, bu da nöbetlere yol açabilir.
Not: Dravet sendromunda SCN1A geni dışında sinirler arası elektrik akımlarını düzenleyen birden fazla gen rol oynar ve bu genlerde meydana gelen mutasyonlar da nöbetlerin ortaya çıkmasında rol oynarlar.
İlaçlı Tedavi
Dravet sendromu bir spektrum bozukluğudur, yani hastalarda çok çeşitli şiddette nöbet tipleri vardır. Bu yüzden birine faydalı olan bir ilaç diğerine faydalı olmayabilir.
Dravet sendromundaki nöbetleri kontrol etmek zordur ancak Antikonvülzan /antiepileptik ilaçlar nöbetleri azaltılabilir. ABD Gıda ve İlaç Dairesi FDA, 2 yaş ve üzeri Dravet sendromlu çocukların kısmi yada generalize istemsiz kasılma sıklığını azaltan ve etken maddesi fenfluramin olan Fintepla adında bir ilacı onayladı ve ilaç şubat 2021 yılında piyasaya çıktı.
Haziran 2018’de ABD Gıda ve İlaç Dairesi, 2 yaş ve üstü Dravet sendromlularda nöbetlerin tedavisi için Epidiolex ve Diacomit’i onaylamıştı. Bazı tedaviler bazı hastalarda tam olarak çalışmasa da bazı hastalarda olumlu sonuçlar vermiştir. IVIG tedavisi (İntravenöz immünglobülin), ketojenik Diyet ve VNS (Vagus Sinir Stimülasyonu) buna örnektir.
Acil durum ilaçları
Birçok hasta, acil müdahale gerektiren uzun süreli nöbetler (status epilepticus) yaşar. Bu nedenle doktorlar nöbet durdurmak için içeriğinde benzodiazepin olan bir kurtarma ilacı reçete edebilir.
Bazı acil durum ilaçları şunlarıdır
- Klonazepam (Klonopin)
- Diazepam (Diastat)
- Lorazepam (Ativan)
- Midazolam (Versed)
Gen Tedavisi
SCN1A geni 6000 nükleotidten oluşan oldukça büyük bir gendir. Genin büyük olması yıllardır uygulanan geleneksel gen tedavi metotları için zorluk oluşturuyordu. Ancak son yıllarda geliştirilen yeni teknikler tedaviyi daha yakın hale getirdi.
Yukarıda da belirtildiği gibi Dravet sendromu vakalarının %90’ından fazlası Nav1.1’i kodlayan SCN1A geninin iki kopyasından birinde meydana gelen mutasyonlardan kaynaklanıyor. Bu da Nav1.1 sodyum kanallarının %50 daha az olması anlamına geliyor.
Şizofreni gelişiminde plasentanın rolü: Annenin yaşadığı stres risk yaratabilir
Gen tedavisine yönelik çalışmalar iki ana strateji üzerine kurgulanmaktadır. Bunlardan biri mutasyonsuz SCN1Agenini daha fazla çalışmaya teşvik etmek yani genin ekspresyonunu artırmak, diğeri ise mutasyonlu geni yeniden dizayn etmek.
Tasarlanmış transkripsiyon faktörü ile tedavi: ETX101
ETX101, Dravet sendromunun sebep olduğu tüm nöbet, bilişsel, davranışsal, gelişimsel ve Motor becerileri geliştirmek için geliştirilmiş hücre seçici bir gen tedavisidir.
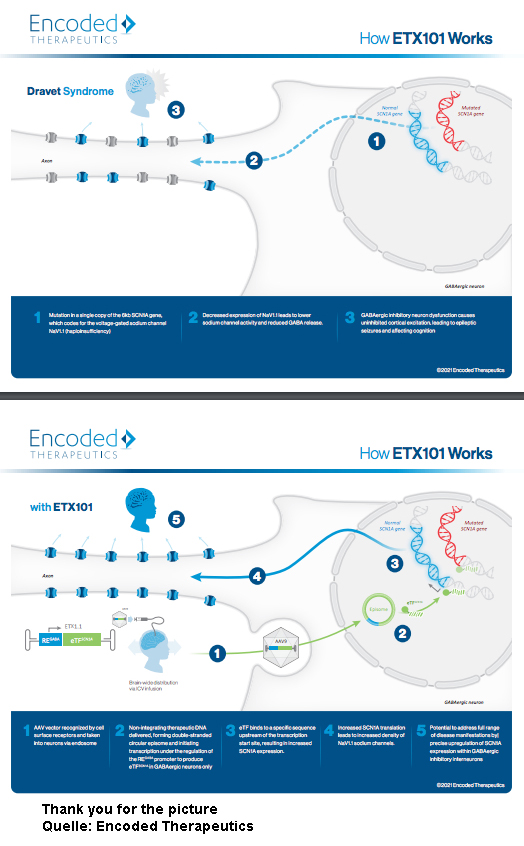
Merkezi San Francisco bulunan Encoding Therapeutics firması geliştirdiği ETX101 adında yeni bir gen terapisi stratejisi ile SCN1A geninin ekspresyonunu artırmayı başardı. ETX101, SCN1A’nin yeni bir kopyasını yapmak yerine, genin sağlıklı kopyasının daha fazla çalışmasını teşvik ediyor (ekspresyonunu yükseltiyor) ve bu sayede sodyum kanalı Nav1.1’nin sayısı artıyor. Sodyum kanalı Nav1.1 artması ise nöronlar arasında iletişimi gerçekleşmesine ve buna bağlı olarak Dravet sendromunun tedavisini mümkün kılıyor. (şimdilik deney hayvanlarında)
Encoding Therapeutics firması yaptığı açıklamada, bu tedavi yaklaşımının Dravet sendromlu fare modellerinde etkili olduğunu belirtiyor. Firma ayrıca ETX101’nin primatların beynine bir virüs vasıtasıyla (Adeno-associated viruses (AAV) enjekte edildiğini ve olumlu sonuçlar verdiğini de belirtiliyor. Encoding Therapeutics firması 2021’in sonunda insanlarda deneme hazırlıklarına başlamayı ve 2022’de ilk hastaları tedavi etmeyi planlıyor.
Körlük ve kök hücre tedavisi: Yeni geliştirilen yöntem körlüğe çare olabilir
CRISPR tekniği ile tedavi
CRISPR, klinik öncesi hücre ve kemirgen modellerinde test edilen bir başka yaklaşım. Bu teknik ile SCN1A geninin düzenlenmesini hedeflemektedir. CRISPR/Cas9 teknolojisi yaygın olarak DNA dizilerinde hatalı kısmı kesmek ve dizayn edilmiş sağlıklı kısmı yapıştırmak şeklinde kullanılır. Bununla birlikte, bazı araştırmacılar bu teknolojiyi gen ekspresyonunu artırmak için farklı bir şekilde kullanmakta. Araştırmacılar bu tür kullanımda kesme işlemini gerçekleştiren Cas9 enzimini devre dışı bırakarak
SCN1A geninin mutasyonsuz kopyasının çalışmasını artırmanın yolunu araştırıyor. Fare modellerinde yapılan çalışmalar, bu yaklaşımın sağlıklı SCN1A genini daha fazla çalıştırarak Nav1.1 sodyum kanalı sayısını arttırdığını gösteriyor.
Antisens oligonükleotidtir (ASO) ile tedavi: STK-001
STK-001, hücrede SCN1A-mRNA seviyesinin artmasını teşvik ederek Nav1.1 sodyum kanalının üretiminin normale yakın seviyelere gelmesini sağlayan bir Antisens oligonükleotidtir (ASO). Antisens oligonükleotit veya ASO olarak adlandırılan STK-001, hücrelere erişmesine izin veren bir lipid damlacığı içinde paketlenebilir ve ardından intratekal enjeksiyon ile (omurilikten) veriliyor.
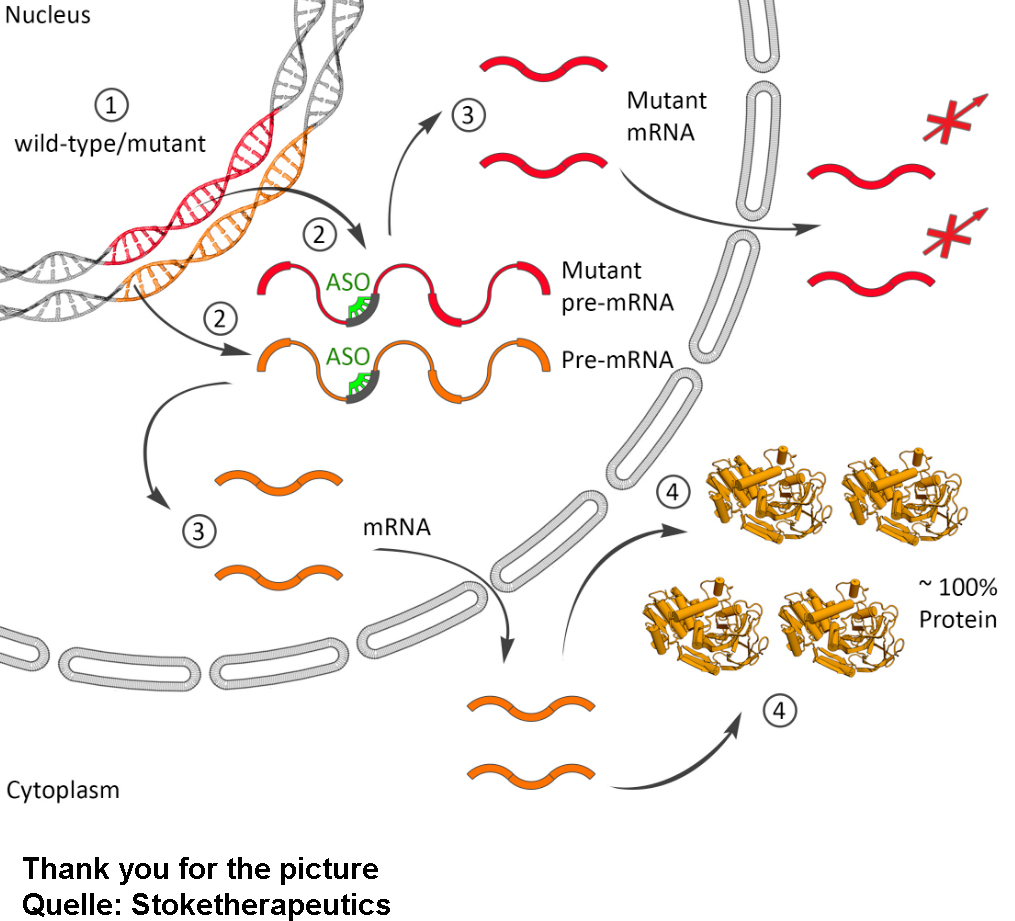
Ön klinik çalışmalar, Dravet sendromlu fare modelinde nöbetleri ve mortaliteyi azaltmada etki gösterdi. STK-001’in Dravet sendromlu hastalarda güvenliğini belirlemek için 2020’nin sonlarında ve 2021’in başlarında klinik denemeler başladı. STK-001 ile yapılan çalışmanın ilk sonuçlarının 2021’in sonuna kadar rapor edilmesi bekleniyor.
Klonlama tekniği ile tedavi
Yukarıda bahsedildiği gibi, Dravet sendromunda gen tedavisi için en büyük zorluklardan biri, SCN1A geninin 6000 nükleotid uzunluğundaki boyutudur. Çünkü bu büyüklükteki bir gen uygun bir vektörlere sığmıyor (klonlanamıyor).
Çeşitli araştırma grupları SCN1A geninin sağlıklı bir kopyasını hücreye sokabilecek daha büyük vektörler üzerinde çalışıyor. Bazı çalışma grupları ise SCN1A genini bölerek iki farklı vektöre klonlama yaparak hücreye sokmayı ve hücreye girdikten sonra tekrar iki parçayı birleştirmeyi deniyor. Bu çalışmaların tümü hala erken aşamada ancak hücre kültürü ve fare modellerinde yapılan bazı çalışmalar umut verici sonuçlar vermeye başladı. Bu yaklaşımların insanlarda kullanmanın zorluklarının ne olabileceği henüz belinmiyor ancak çalışmalar istikrarlı bir şekilde ilerliyor.
Multipl skleroz (MS) nedenleri ve tedavisindeki yeni bilimsel gelişmeler!
Öngörü ve tavsiyeler
Nöbet tetikleyicilerden kaçınılmalı
Mümkün olduğunca nöbet tetikleyicilerden kaçınılmalı. Dravet sendromlu hastalar için yaygın tetikleyiciler arasında çevre ve/veya vücut sıcaklığındaki hızlı değişiklikler ile hastalıklar, stres, aşırı heyecan, desenler ve yanıp sönen ışıklar. Aileler çocuklarını nöbet tetikleyici durum ve ortamlardan uzak tutmalı ve bunun için özel camlı gözlükler ile serin tutan giysiler sağlanmalı.
Yüksek Ateşten kaçınılmalı
Ateşin yükselmesi durumunda bir çocuk doktoru veya nörolog tarafından bir plan dahilinde acilen tedavi edilmeli.
Diğer zorluklar
Dravet sendromlu çocuklar genellikle bilişsel gecikmeler, iletişimde zorluklar, sosyal beceri ve davranışlarda sorunlar gibi otizmli bireylerde görülen benzer semptomlar sergileyebilirler. Bu yüzden hastalara düzenli gelişimsel değerlendirmeler yapılarak konuşma ve gelişim konusunda destek verilmeli.
Aile olarak başa çıkmak
Gerek Dravet sendromlu gerekse diğer kronik hastalığa sahip çocukların aileleri büyük bir yük ve sorumluluk altındadır. Bu durum zaman zaman öfke, korku, şok, kafa karışıklığı, kendini suçlama ve çaresizlik gibi ruhsal tepkimeler verilmesine sebep olur. Bu yüzden aileler bir aile danışmanından destek almalı ve aynı durumdaki aileler ile dayanışma içerisinde olmalıdırlar.
Dravet sendromlu çocuklar yaşlandıkça, bilişsel işlevlerdeki düşüş dengelenir. Yaşla birlikte atakların sayısı ve süresi azalır. Yürüyüş anormallikleri ergenlik döneminde daha da kötüleşiyor gibi görünüyor. Dravet sendromlu kişilerde ani, beklenmedik ölüm riski genel popülasyondan daha fazladır, ancak bu risk hala küçüktür.
Benzer konularda hazırlanmış diğer makaleler
- Gen terapi, epilepsi hastaları için yeni bir umut
- Otizmin bazı formları gelecekte ilaçla tedavi edilebilir
- Genetik Hastalıklar: Osteogenezis imperfekta (OI)
- Genetik hastlıklar: Hemofili
- Genetik hastalıklar: Huntington
- Kromozomal hastalıklar-1 (Trizomi 21, Trizomi 18 ve Trizomi 13)
Mehmet Saltuerk
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics of the University of Cologne
++++++++++++++++++++++++++
Kaynaklar
- Incidence of Dravet Syndrome in a US Population
- Mortality in Dravet Syndrome
- Dravet syndrome and parent associations: The IDEA League experience with comorbid conditions, mortality, management, adaptation, and grief
- SCN1A Seizure Disorders
- Migraine Genetics: Part II
- 1 channels and epilepsy
- PDF Figures Save Share Reprints Request dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice
- Antisense oligonucleotides increase Scn1a expression and reduce seizures and SUDEP incidence in a mouse model of Dravet syndrome
- Antisense oligonucleotide modulation of non-productive alternative splicing upregulates gene expression
- Incidence of Dravet Syndrome in a US Population
- CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice
- Genotype–phenotype associations in SCN1A-related epilepsies
YAZIYI PAYLAŞ
Yazar Hakkında
Mehmet Saltuerk
Genetik
Benzer Yazılar
 Down sendromu nedir, neden olur? Belirtileri ve tedavisi
Down sendromu nedir, neden olur? Belirtileri ve tedavisi- Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
- Sodyum eksikliği ve yüksekliği neden olur? Belirtileri ve tedavisi
- Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi
- Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
 Down sendromu nedir, neden olur? Belirtileri ve tedavisi
Down sendromu nedir, neden olur? Belirtileri ve tedavisi Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler Sodyum eksikliği ve yüksekliği neden olur? Belirtileri ve tedavisi
Sodyum eksikliği ve yüksekliği neden olur? Belirtileri ve tedavisi Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi
Kromozomal hastalıklar (Trizomi 21-18-13) ve Down sendromu, tanı ve tedavisi Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
YORUMUNUZ VAR MI?
Tedavi bulunmadı mi hâlâ gerçekten buna ihtiyacımız var yıl 2023
Dravet sendromu tedavisi icin araştırmalar fareler üzerinde etkili olduğu söylenilmis ve 2021in sonunda sunulacaği yada olumlu olup olmadıği açiklanacaği yazılmıs 2023 yılına girdik ve tıp artık cok ilerledi lütfen bulun bu tedaviyi gerçekten ailelerin buna çok ihtiyacı var peki bu araştırma şuan.ne durumda