
Genetik Hastalıklar: Osteogenezis imperfekta (OI) nedenleri, belirtileri ve tedavisi

Tanım: Halk arasında “cam kemik hastalığı” olarak adlandırılan Osteogenezis imperfekta (OI), nadir görülen genetik bir hastalıktır. Hastalık çeşitli genlerde meydana gelen mutasyonlardan kaynaklanır. Bu mutasyonlar kollajen adında bir proteinin yanlış sentezlenmesine yol açar. Hastalar yapısı bozulan bu protein nedeniyle başta ciddi kemik kırılmaları olmak üzere hayat boyu yaşamı kısıtlayıcı birçok semptomla karşı karşıya kalabilirler.
Hastalarının kemikleri görünürde bir neden olmaksızın veya hafif travmada kolayca kırılabilir. Birden fazla kırıklar yaygındır hatta doğumdan önce bile ciddi kırık vakaları olabilir. Hafif vakalarda ise yaşam boyunca yalnızca birkaç kırık olabilir. Osteogenezis imperfekta, çoğunlukla kemiklerleri etkilemesine rağmen dişler, gözler, kulaklar ve iç organlar da bu hastalıktan etkilenebilir.
Osteogenezis imperfekta – Epidemiyoloji
Hastalığın toplumda görülme sıklığının 1: 1.000 ile 1: 30.000 arasında olduğunu gösteren farklı görüşler vardır. Literatürler bu farklı frekans bilgisi için hafif formlarının geç teşhis edilmesini gösteriyor. Bu durum Osteogenezis imperfektanın toplumda ne sıklıkta olduğunu belirlemesini güçleştiriyor.
Genetik Hastalıklar: Ehlers Danlos Sendrom(EDS) nedir? Belirtileri ve tedavisi
Osteogenezis imperfekta – Nedenleri
OI, genetik olarak heterojen bir hastalık grubudur ve nadir görülen özel formları vardır. Hastalık, hem otozomal dominant, hem de otozomal resesif olarak miras alınır. En hafif görülen Tip I formu ile orta ve şiddetli formların % 77’si COL1A1 ve COL1A2 genlerindeki mutasyonlardan kaynaklanır. Orta ve şiddetli vakaların yaklaşık % 9’unun sebebi ise IFITM5 genindeki mutasyonlar dır. Klinik semptomların şiddeti mutasyonun türü ve hangi gende bulunduğuna bağlı olarak değişir.
Açıklama: COL1A1 ve COL1A2 genleri, Tip I kollajen proteini sentezlemek için gerekli talimatları içerirler. Bu protein, deri, kemik ve diğer bağ dokularında bolca bulunarak vücudun sağlam güçlü kalmasını sağlar. Osteogenezis imperfekta Tip I, genellikle COL1A1 genindeki ve daha az olarak da COL1A2 genindeki mutasyonlardan kaynaklanır. Bu mutasyonlar üretilen Tip I kollajen proteinin yapısını bozmaz ama miktarını azaltır, bu da bağ dokuları, özellikle kemikleri zayıflatır.
Diğer genlerdeki mutasyonlar nadir görülen Osteogenezis imperfekta’nın diğer formlarına neden olur. Bu genlerin bir kısmı regülatör genlerdir. Regülatör genler Tip I kollajeni senteleyen geni regüle ederek Tip I kollajenin olgun forma dönüşmesine yardımcı olurlar. Bu genlerdeki mutasyonlar, Tip I kollajenin üretimindeki farklı aşamaları bozar. Bu değişiklikler bağ dokularını zayıflatarak ciddi kemik anormalliklerine ve büyüme sorunlarına yol açarlar.
Genetik Hastalıklar: Charcot Marie Tooth (CMT); belirtileri, tipleri ve tedavisi
OI’nin nadir görülen otozomal resesif kalıtımsal özel formları çoğunlukla spesifik klinik özelliklerle karakterizedir. Bu genlerin analizi kapsamlı klinik değerlendirmeler ile yapılır.
Osteogenezis imperfektanın semptomları
Osteogenezis imperfekta nın (OI) semptomları, formlar arasında farklılıklar gösterir. Bu yüzden genel semptomlar çok hafiften şiddetliye kadar değişebilir. Öyleki hastalığın şiddeti aynı ailenin hasta bireyler arasında bile farklılıklar gösterebilir. Osteogenezis imperfektanın ana semptomu kırık kemiklerdir. Kemikler çok kolay kırılabilir, hatta bebeklerde bez değiştirirken veya geğirirken bile kemiklerin kırılması mümkündür.

Belirgin semptomlar:
- Kolayca kırılan kemikler
- Kemik deformiteleri ve bacakların fleksiyonunda sorunlar
- Bacakların eğilmesi ve skolyoz adı verilen kavisli omurga
- Varil şeklinde bir göğüs
- Eğimli bir sırt
- Gevşek eklemler (Eklemler hipermobil veya çok esnek olabilir
- Bir dereceye kadar eklem veya kemik ağrısı olabilir
- Zayıf kaslar ve dokular
- Etkilenenlerin çoğunda kas gerginliği
- Kolayca zedelenen cilt
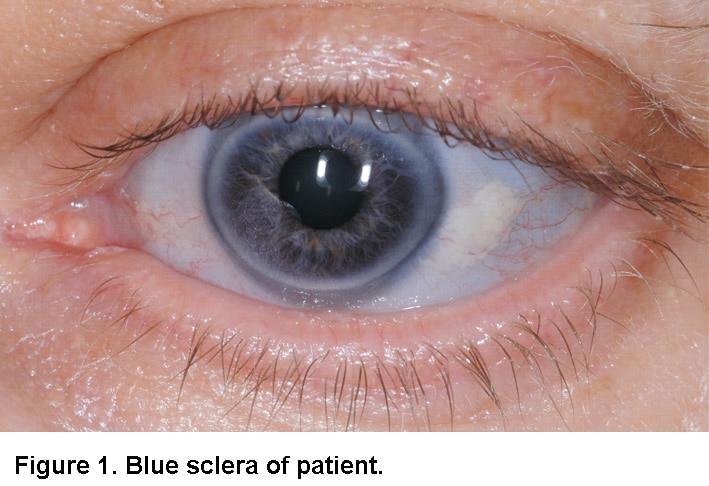
- Göz beyazlarında (sklera) renk değişikliği. Mavi veya gri olabilir. Özellikle Tip I ve VI gibi hafif formlarda daha yaygındır
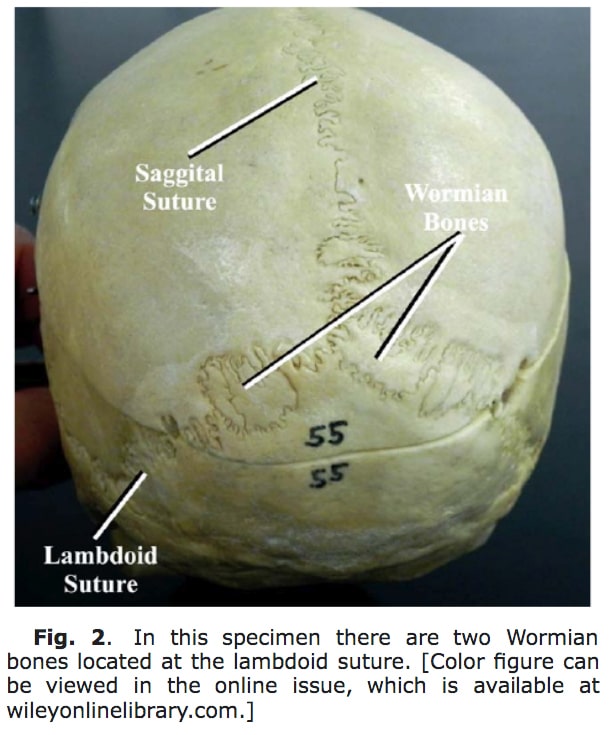
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Erken yetişkinlikte işitme kaybı
- Üçgen yüz
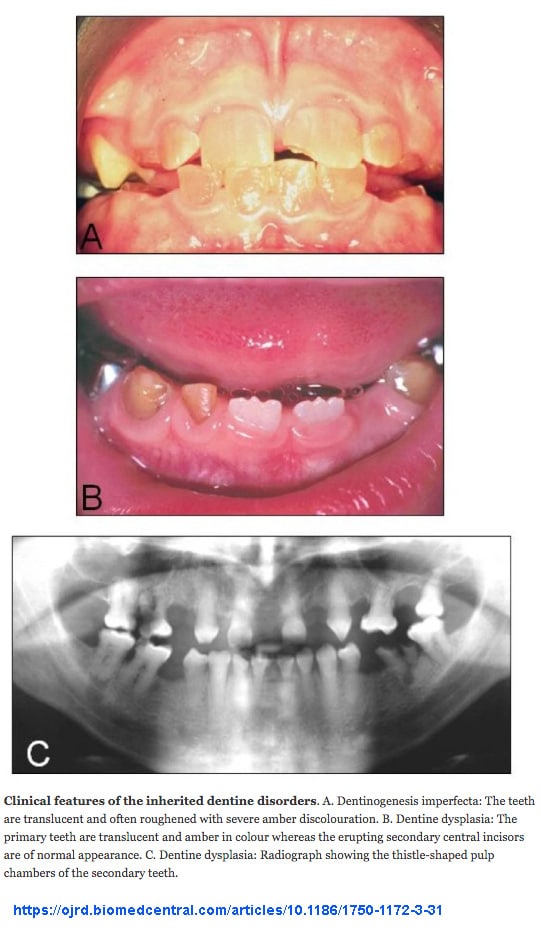
- Diş oluşumuyla ilgili sorunlar: Yumuşak, rengi bozulmuş dişler
- Solunum Problemleri
- Aşırı yorgunluk hissi
- Kolayca yaralann cilt
- Kanama ve kolay morarma (Sık burun kanaması veya bir yaralanmadan sonra ağır kanama)
- Erken yetişkinlikte başlayan işitme kaybı
- Sıcak havaya karşı dayanıksızlık
- Vücut ısısı nın genellikle yaklaşık 0,5 ° C daha fazla olması. (Bilimsel olarak bu fenomen hala açıklanamamıştır. Bu aynı zamanda daha kolay terleme eğilimiyle de ilişkilidir)
- Kısa boy
Hafif formdaki semptomlar
- Belirtiler daha azdır
- Kemik deformitesi çok az veya hiç yok
- Kırık kemik sayısı ve kırılma sıklığı daha az
- Boy, genellikle etkilenmez
- Erken işitme kaybı olabilir
- Kemik kırılmaları ergenlikten sonra azalır
- Ortalama bir yaşam beklentisi
Osteogenezis imperfektanın semptomları diğer hastalıklara benzeyebilir, bu yüzden kesin teşhis için daima doktor ile iletişime geçilmelidir. (17)(18)(19)

Osteogenesis imperfektanın (OI) sınıflandırması
Osteogenesis imperfektanın (OI) Tip I den Tip XIX a kadar bilinen 19 formu vardır. Aşağıda Osteogenesis imperfektanın on dokuz formundan sekizine yer verilmiştir. Ayırt edici özellikleri örtüşse de bazı türler belirti ve semptomlar bakımından farklılık gösterir.
OI Tip I (eski adıyla Osteogenesis imperfecta tarda, tip Lobstein): Genellikle 17. kromozom bulunan COL1A1 veya 7. kromozomda bulunan COL1A2 genlerindeki heterozigot mutasyondan kaynaklanır. Bu mutasyonlar Tip I kollajen miktarlarının azalmasına yol açar.
Kromozomal Hastalıklar – 2: Angelman Sendromu ve Prader-Willi Sendromu
Tip I, OI’nin en hafif formudur. Genellikle çocuklar yürümeyi öğrenirken kırıklar oluşmaya başladığında fark edilir. Bu formdan etkilenen kişiler genellikle yaşamın üçüncü on yılının ortalarına kadar teşhis edilmezler. Yenidoğan çocuklarda kırıklar nadirdir. Kırık eğilimi çocukluktan ergenliğe kadar sabittir, daha sonra azalır. Kırıklar sıklıkla kadınlarda menopozdan sonra ve erkeklerde altıncı on yıldan sonra artar.
İşitme kaybı, genellikle ergenlik çağında veya genç yetişkinlik döneminde başlayabilir, ancak daha erken de başlayabilir. Tip I OI’li bazı hastalar bu hastalıktan çok hafif etkilenir. Bu gruptaki kişiler, ortalama yürüyebilir ve koşabilir ve gevşek eklemler gibi zar zor farkedilebilen hafif belirtilere sahiptirler. Bu durumdaki hastalar hastalıktan o kadar hafif etkilenir ki, gençlik veya yetişkinlik yıllarına kadar teşhis edilemezler. (1)(2)(14)
Yaygın semptomlar:
- Normale yakın boy (Boy, genellikle hastalıktan etkilenmemiş aile üyelerinden daha kısadır)
- Yetişkinlik döneminde işitme kaybı. (Kadınlarda erkeklere göre iki kat fazla)
- Ailelerin yaklaşık % 50’sinde, onlu yaşların sonlarından başlayarak ve dördüncü ila beşinci on yılın sonunda kademeli olarak derin sağırlık
- Gözün beyaz kısmının mavileşmesi (mavi sklera)
- Çoğu hastada dişler normal (nadir Dentinogenezis imperfekta)
- Kalpde mitral kapak prolapsusu (Kalpteki bir ya da iki kapakçığının kalbin kasılması sırasında sol kulakçığa doğru bombeleşmesi, kubbeleşmesi ya da çökmesi)
- İskelette hafif osteopeni (Düşük kemik yoğunluğu)
- Değişen derecelerde çoklu kırıklar
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Hafif ve orta derecede eklem gevşekliği (eklem hipermobilitesi) ve kifoskolyoz (omurga eğriliği)
- Ara sıra uyluk kemiğinde eğilme (femoral eğilme)
- İnce ve kolay moraran cilt
- Biconcave düzleştirilmiş omurlar (Her iki yüzeyi de içbükey veya oyuk olan)
- Çocuk yürümeye başladığından itibaren sıklıkla ama ergenlikten sonra azalan kırı (Kırıklar genellikle deformite olmaksızın iyileşir)
- Kadınlarda menopoz sonrası, erkeklerde 60-80 yaş arası kırılma sıklığı artar
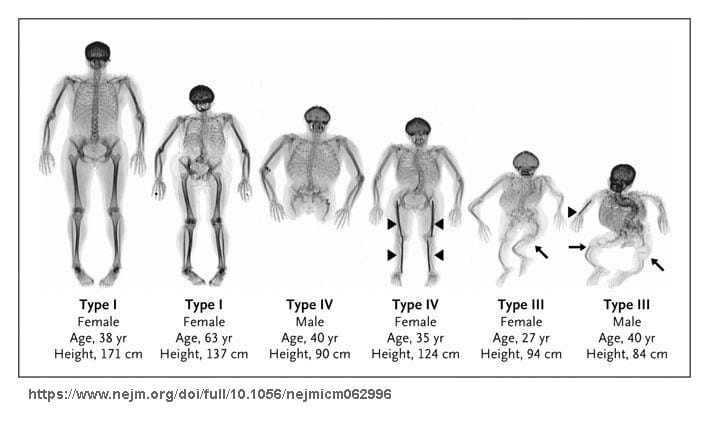
OI Tip II: COL1A1 veya COL1A2 genlerindeki heterozigot mutasyondan kaynaklanır. Tip II, OI vakalarının yaklaşık % 20’sinde görülen en şiddetli formdur. Perinatal dönemde birçok perinatal kırık, uzun kemiklerde şiddetli eğilme ve kemik kırılganlığı ile karakterizedir. OI Tip II ile doğan bebeklerin % 60’ı gelişmemiş akciğer fonksiyonu nedeniyle ilk birkaç hafta içinde ölürler. Bir yıldan fazla hayatta kalmaları nadirdir. OI Tip II, otozomal dominant modelde kalıtılır.(3)
Yaygın semptomlar:
- Anne karnında bebekte su toplama (İmmün olmayan hidrops)
- Erken doğum
- Düşük doğum ağırlığı
- Kısa uzuvlar ve cücelik
- Gözün beyaz kısmının mavileşmesi (mavi sklera)
- Gagalı burun
- Konjestif kalp yetmezliği (Kalp kaslarının yeteri kadar kan pompalayamaması sonucu oluşan kalp yetmezliği)
- Akciğer yetmezliği
- Doğumda var olan çok sayıda çoklu kırık
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Geniş buruşuk uzun kemikler
- İnce deri
- Geniş buruşuk uzun kemikler
- İç içe geçmiş uyluk kemiği
- Kalçalar ve ayaklarda deformasyonlar
- Omurganın düzleşmesi (Platyspondyly)
- Kafatasının üst kısmı yumuşaklık (yumuşak calvaria)
- Büyük fontaneller (kafatasının ön-üst kısmı)
- Düşük kalsiyum ve fosfor düzeyi (Kalvaryal mineralizasyon)
- Tibial eğilme (Diz kıkırdağının tahribatına bağlı olarak bacak kemiğinde iç tarafa doğru eğilme)
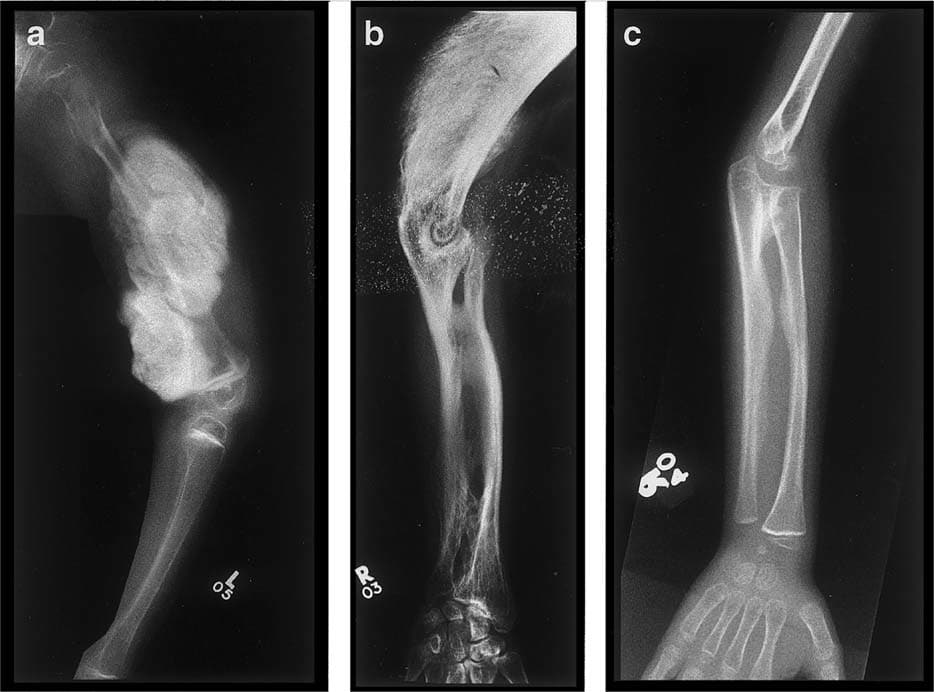
OI Tip III: (Eski adıyla osteogenesis imperfecta congenita, tip Vrolik): COL1A1 veya COL1A2 genlerinden birinde meydana gelen heterozigot mutasyon dan kaynaklanır. Doğum sürecinde sıklıkla çoklu kırıklar meydana gelir. Ergenliğe kadar yaklaşık 100 kemik kırılması gerçekleşir. Fenotip özellikler değişkendir. Tipik olarak son derece kısa boy, ince, kavisli ve yumuşak kemikler, iskelet deformiteleri, skolyoz, dentinogenezis imperfekta ve işitme kaybı ile karakteristiktir. OI Tip III tüm osteogenesis imperfecta vakaların yaklaşık % 5 ni oluşturur. OI Tip III, otozomal dominant modelde kalıtılır. (4)(20)
Kromozomal hastalıklar ve gebelik: 35 yaş üstü hamilelikler neden riskli
Yaygın semptomlar:
- Kısa uzuvlar, cücelik. Yetişkinlikte boy 92-108 cm
- İnce üçgen yüz (Triangular face)
- Normalden küçük çene (Mikrognati)
- Alın ve kaş bölgesinde öne doğru çıkıklık (Frontal bossing)
- İşitme kaybı
- Gözün beyaz kısmının mavileşmesi (mavi sklera). (Yaşla birlikte normale döner)
- Dentinogenesis imperfecta (hem birincil hem de ikincil dişler)
- Akciğer atardamarında sürekli yüksek kan basıncı (Pulmoner hipertansiyon)
- İnce kaburgalar
- İskelette şiddetli osteoporoz ve doğumda birden fazla kırık
- Büyük ön fontanel
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Düşük Kalsiyum ve fosfor düzeyi (Kalvaryal mineralizasyon)
- Omurgalarda skolyoz, kifoz
- Özellikle bel omurlarda iç bükey deformasyonlar(Codfish vertebrae)
- Kalça eklemi deformiteleri (Protrüzyo Asetabulinin)
- Doğumda veya yaşamın ilk 2 yılında ortaya çıkan uzun kemik deformiteleri
- Birden fazla kırık nedeniyle uzuvların eğilmesi
- İnce uzun kemikler
- Tibial eğilme (Diz kıkırdağının tahribatına bağlı olarak bacak kemiğinde iç taraf doğru eğilme)
- Kısa deforme olmuş femurlar
OI Tip IV: COL1A1 veya COL1A2 genlerindeki heterozigot mutasyondan kaynaklanır. OI Tip IV, OI’nin orta dereceli bir formudur. Hastalıktan etkilenenlerin yaklaşık yarısında işitme kaybı vardır. Sklera genellikle beyazdır ya da hafif mavimsi dir. Bu formdakiler de kısa boyludur ve kemiklerde sadece hafif deformasyonlara vardır. Kemiklerin kırılma ve deformasyon eğilimi Tip III e göre daha azdır. Tekerlekli sandalye gerekli değildir. OI IV, otozomal dominant modelde kalıtılır.(6)(7)
Yaygın semptomlar:
- Kısa boy
- İşitme kaybı
- Orta ve iç kulakta anormal kemik yapısı (Otoskleroz)
- Gözün beyaz kısmının mavileşmesi (mavi sklera). Soluk mavi skleralar (vakaların% 10’u)
- Dişlerde Dentinogenezis imperfekta
- İskelette değişken derecelerde çoklu kırıklar.
- Hafif-orta derecede iskelet deformitesi
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Omurgalarda skolyoz kifoz
- Biconcave vertebra (Düzleşmiş omurlar: her iki yüzeyi de içbükey veya oyuk)
- Uzuvlarda doğumda mevcut olan femoral eğilme, zamanla düzelme
- Birden fazla kırık nedeniyle eğilmiş uzuvlar
- Doğum sancıları sırasında veya doğumdan sonraki dönemde ortaya çıkabilen kırıklar
- Ergenlikten sonra azalan ancak menopozdan sonra artan kırıklar
- Hafif-orta derecede iskelet deformitesi ve değişken derecelerde çoklu kırıklar
OI Tip V: 11. kromozomda bulunan IFITM5 genindeki heterozigot mutasyondan kaynaklanır. OI Tip V, orta şiddettedir ve görünüm ve semptomlar açısından Tip IV’e benzer. Belirgin özellikler arasında kırık veya cerrahi bölgelerde oluşabilen hipertrofik nasırlar ve radius ile ulna (ön koldaki 2 kemik) arasındaki zarın kireçlenmesi nedeniyle sınırlı önkol hareketi. OI V, otozomal dominant modelde kalıtılır.(5)(8)(9)
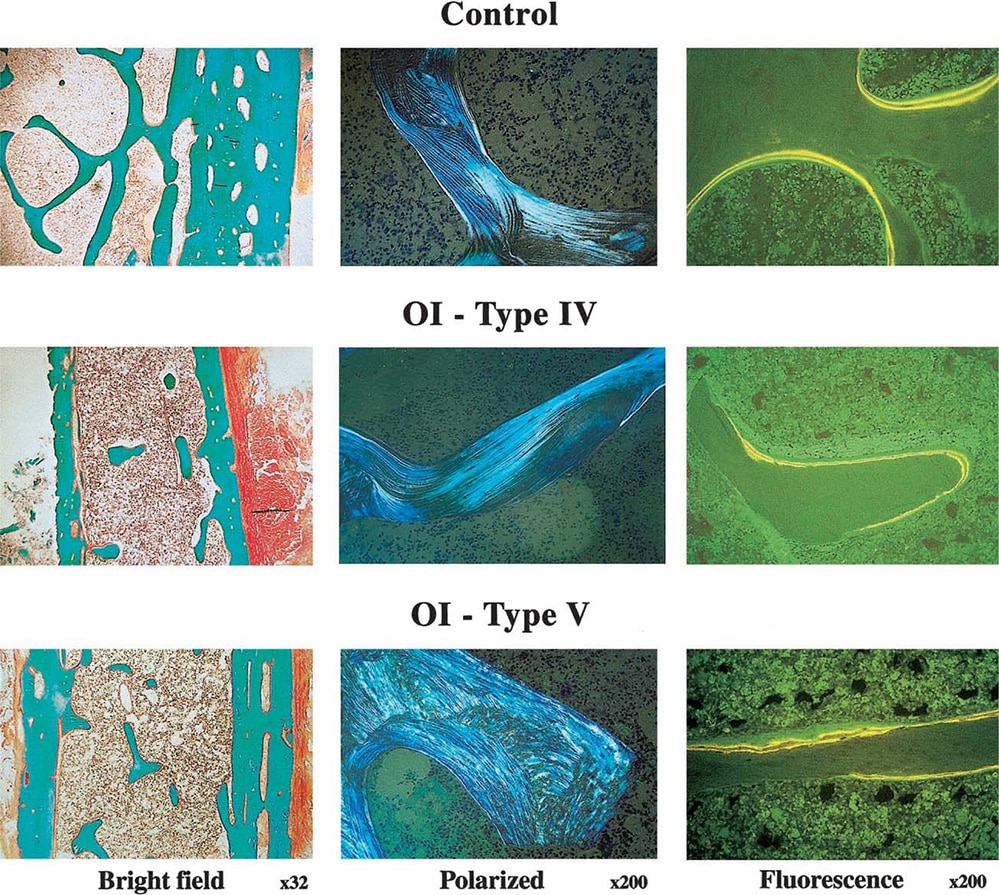
Yaygın semptomlar:
- Doğum uzunluğu normal
- Çocuklukta kısa boy
- Doğum ağırlığı normal
- Üçgen yüz
- Gözün beyaz kısmının mavileşmesi (mavi sklera).
- Dişlerde nadir olarak Dentinogenezis imperfekta
- Nadir olarak dişlerin kesme yüzeyinde belirgin, düzensiz çıkıntılar
- İskelette orta derecede deforme olan osteogenezis imperfekta
- Değişken derecelerde çoklu kırıklar
- İskelette düşük kalsiyum ve fosfor düzeyi (Kalvaryal mineralizasyon)
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Biconcave vertebra (Düzleşmiş omurlar: her iki yüzeyi de içbükey veya oyuk)
- Nadir olarak transiliak biyopside mineralize edilmemiş geniş osteoid bantları
- Kama şeklindeki omurlar
- Önkolun sınırlı dönüşü (pronasyonu / supinasyonu)
- Dirsek ve büyük eklemlerde çıkıklar (Radyal başın anterior çıkığı)
- Radius ile ulna (ön koldaki 2 kemik) arasındaki zarın kireçlenmesi nedeniyle sınırlı önkol hareketi
- Hiperplastik nasır
- Aşırı uzayabilen eklemler (bazı hastalarda)
- Eğer aile içinden birden fazla OI Tip 5 varsa aile bireyleri arasında değişen fenotipler
- Aynı mutasyonu taşıyan hastalar arasında bile oldukça değişken derecede kemik kırılganlığı
- Doğum öncesi yapılan ultrasonlarda kemik anomalileri görülebilir (bazı hastalarda)
OI Tip VI: 17. kromozomda bulunan SERPINF1 genindeki homozigot mutasyondan kaynaklanır. Oldukça nadir görülen bir formdur ve kemik kırılganlığı ve düşük kemik kütlesi ile karakteristiktir. Belirtiler orta düzeydedir. Tip IV’e benzer. Yapılan kemik Biyopsisinde karakteristik bir mineralizasyon kusuru ile ayırt edilir. Sklera normalden hafif mavimsi dir ve Dentinogenesis imperfecta yoktur. OI VI, otozomal resesif modelde kalıtılır. (10) (11)
Yaygın semptomlar:
- Doğum öncesi kırıklar olabilir de olmayabilir de
- Doğum sonrası kısa boy,
- Gözün beyaz kısmının mavileşmesi (mavi sklera).
- Eklem hipermobilitesi (bazı hastalarda)
- Hafif kemik yaşı gecikmesi,
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Yumuşak kafatası (bazı hastalarda)
- Omurgalarda, skolyoz, kifoz
- Gecikmiş kalça gelişimi
- Leğen kemiğinde, küçük pelvis ve gecikmiş kalça gelişimi
- Uzun kemiklerde kırıklar (üst ve alt)
- Eğik uyluk kemiği
- Eğik omuzlar
- Üst ekstremitelerde eğilme
- Alt ekstremitelerde eğilme
- Üst ekstremite kısalık
- Alt ekstremitelerin kısalık
- Kol ve bacak gibi büyük kas gruplarını kontrol etme yeteneğinde gecikme
- Değişken şiddette kırık
- İskelette Osteopeni, Kırıklar, Eklem hipermobilitesi (bazı hastalarda)
OI Tip VII: 3. kromozomda bulunan CRTAP genindeki homozigot veya bileşik heterozigot mutasyondan kaynaklanır. OI Tip VII, Osteogenezis imperfekta nin şiddetli veya öldürücü formudur. Bu form nadir bir formdur ve tüm imperfekta vakalarının % 2 ila 3’ünü oluşturduğu tahmin ediliyor. Çoklu kemik kırıkları, aşırı derecede düşük mineralizasyon ile karakterizedir. OI VII, otozomal resesif modelde kalıtılır. (12)
- Yaygın ters doğum ((Breech presentation)
- Normal doğum uzunluğu ama yetişkinlikte kısa boy
- Normal doğum ağırlığı
- Kafatası: ön fontanelde büyük açık
- Kafatası: Arka suturede açık
- Yuvarlak yüz
- Uzun philtrum (üst dudak ile burun arasındaki mesafe)
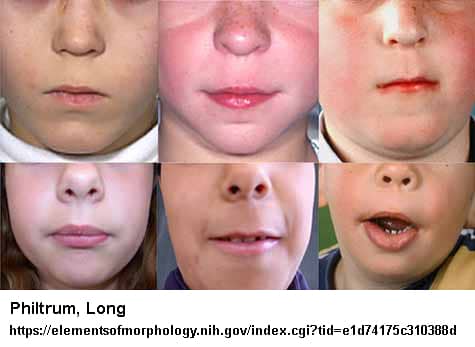
- Normal işitme
- Gözün beyaz kısmının mavileşmesi (mavi sklera).
- Proptozis (gözlerin birbirine göre farklı konumlanması)
- Dişlerde Dentinogenezis imperfekta yok
- Kanı kalpten akciğerlere taşıyan kan damarları yok (Pulmoner arter yokluğu)
- Az gelişmiş pulmoner damarlar
- Göğüs duvarı yapısal bozukluk (Pektus ekskavatum)
- Solunum yetmezliği / pnömoniye sekonder bebeklik döneminde ölüm
- işitme normal
- Dar göğüs
- Çoklu kaburga kırıkları
- Böbrek büyümesi (Hidronefroz)
- Doğumda birden fazla kırık var
- Orta şiddette kemik kırılganlığı
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Omurgada kompresyon kırıkları
- Skolyoz
- Kalça kemiğinde, uzuvlarda anomaliler
- Buruşuk uzun kemikler
- Eğimli alt uzuvlar
- El veya ayaklar normalden daha kısa ve küçük (Micromelia)
- Osteopenik uzun kemikler
- Doğumda birden fazla kırıklar
- Ergenlik sonrası kırılmalarda azalma
- Kemik yoğunluğu azlığı (Osteopeni)
OI Tip VIII: 1. kromozomda bulunan LEPRE1 genindeki homozigot veya bileşik heterozigot mutasyondan kaynaklanır. Tip VIII, İrlandalı göçmenler ve Batı Afrikalılar arasında daha yaygındır. Beyaz sklera, uzun eller ve aşırı derecede zayıflamış kemiklerle karakterizedir. OI VIII, otozomal resesif modelde kalıtılır.
Yaygın semptomlar:
- Orantısız kısa boy
- Kollar kısa ve cücelik
- Geniş açık ön fontanel
- Yumuşak kafatası
- Kafatası kemikleri arasında açıklıklar (Open sutures)
- Yuvarlak yüz
- Gözler normal (Beyaz skleralar)
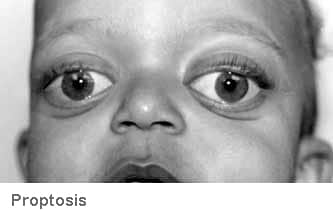
- Proptozis (Gözlerin birbirine göre farklı konumlanması)
- Dişlerde Dentinogenezis imperfekta yok
- İnce kaburga
- Varil şeklinde bir göğüs
- Kırılgan kemikler
- Şiddetli osteopeni
- Normal kemik yaşı
- Doğumda çoklu kırıklar
- Eklem gevşekliği
- Kafatasının üstünde çok sayıda ada benzeri geçişli kemikler (Wormian bones)
- Omurgalarda skolyoz, kifoz
- Platyspondyly Skolyoz
- Omurgalarda kompresyon kırıkları
- İnce, uzun kemikler
- Dışarıya dönük bacaklar
- Uzun falankslar (el ön kemikleri)
- Kısa metakarplar (el arka kemikleri)
- Merkezi sinir sisteminde gelişim gecikmesi
Osteogenezis imperfekta tedavisi
Osteogenezis imperfekta nın şimdilik bir tedavisi yok, ancak semptomlar hafifletebilir, kemiklerin kırılması önlenebilir ve hareket yeteneği en üst düzeye çıkarılabilir. Hastalığın şiddetli formları göğüs kafesi ve omurganın şeklini etkileyerek yaşamı tehdit eden solunum problemlerine yol açabilir. Böyle durumlarda bazı hastaların oksijen takviyesi alması gerekebilir. Ancak birçok vakada hastalar düzenli izleme ve doğru tedavi ile sağlıklı ve üretken bir yaşam sürebilir.
Kişiden kişiye değişen bu tedavilerin optimal yaşam kalitesi sağlayabilmesi için erken müdahale önemlidir. Bu özel tedaviler doktor tarafından belirlenecek bir prosedüre göre yapmalıdır. (15) (16) (21) (22)
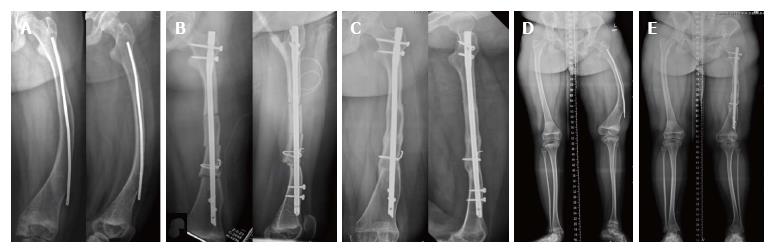
Semptomları önlemeye veya düzeltmeye yönelik tedaviler
- Kırıkların tedavisi (Kırık kemikler için atel ve alçılar)
- Zayıf bacaklar, ayak bilekleri, dizler ve bilekler için destek
- Vücudu güçlendirmek ve hareketleri iyileştirmek için fizik tedavi
- Kollara veya bacaklara metal çubuk yerleştirme (Rodding)
- Kemikleri güçlendiren ilaçlar
- Diş tedavileri (kırılgan dişler için kron gibi özel kaplamalar)
- Özel yapım ekipmanlar (Tekerlekli sandalyeler, diş telleri ve yardımcı cihazlar)
Yararlı olabilecek diğer önlemler
- Normal kiloda kalınmalı (Fazla kilo kemiklere stres verir)
- Doktor kontrolünde güvenli egzersiz yapılmalı
- D vitamini ve kalsiyum açısından zengin gıdalar tüketilmeli (Ancak yüksek doz tavsiye edilmemektedir.)
- Fazla alkolden kaçınılmalı veya sadece ara sıra içilmeli
- Kafein azaltılmalı
- Steroid ilaçların kullanımını doktor tavsiyesine uygun yapılmalı. (Bu ilaçlar kemik
- yoğunluğunu azaltır.)
- Sigara içmekten kesinlikle kaçınılmalı
Benzer konularda hazırlanmış diğer makaleler
- Genetik Hastalıklar: Ehlers Danlos Syndrom (EDS)
- Genetik Hastalıklar: Charcot-Marie-Tooth (CMT)
- Genetik hastlıklar: Hemofili
- Genetik hastalıklar: Huntington
- Kromozomal hastalıklar-1 (Trizomi 21, Trizomi 18 ve Trizomi 13)
- Kromozomal Hastalıklar–2 (Angelman Sendromu, Prader-Willi Sendromu)
- Kromozomal Hastalıklar: 35 yaş üstü hamilelikler neden riskli ?
- Eşcinsellikle ilgili iki mutasyon keşfedildi
- Genler ve hastalıklar
Mehmet Saltuerk
Dipl. Biologe Mehmet Saltürk
The Institute for Genetics of the University of Cologne
++++++++++++++++++++++++
Kaynaklar
Natural history of blue sclerae in osteogenesis imperfecta
Cardiovascular involvement in osteogenesis imperfecta
Deficiency of Cartilage-Associated Protein in Recessive Lethal Osteogenesis Imperfecta
Osteogenesis imperfecta type III with intracranial hemorrhage and brachydactyly associated with mutations in exon 49 of COL1A2
Homozygosity by descent for a COL1A2 mutation in two sibs with severe osteogenesis imperfecta and mild clinical expression in the heterozygotes
A new look at osteogenesis imperfecta. A clinical, radiological and biochemical study of forty-two patients
Genetic heterogeneity in osteogenesis imperfecta
Type V osteogenesis imperfecta: a new form of brittle bone disease
Phenotypic variability of osteogenesis imperfecta type V caused by an IFITM5 mutation.
Osteogenesis imperfecta type VI: a form of brittle bone disease with a mineralization defect
Exome sequencing identifies truncating mutations in human SERPINF1 in autosomal-recessive osteogenesis imperfecta
Deficiency of cartilage-associated protein in recessive lethal osteogenesis imperfecta.
Genetic heterogeneity in osteogenesis imperfecta.
Natural history of blue sclerae in osteogenesis imperfecta
Bone: Use of bisphosphonates in children-proceed with caution
Management of genetic syndromes
Hereditary dentine disorders: dentinogenesis imperfecta and dentine dysplasia
A Case of Spontaneous Intestinal Perforation in Osteogenesis Imperfecta
Wormian bones: A review
Adults with Osteogenesis Imperfecta
Smoking and Bone Health
Using humeral nail for surgical reconstruction of femur in adolescents with osteogenesis imperfecta
YAZIYI PAYLAŞ
Yazar Hakkında
Mehmet Saltuerk
Genetik
Benzer Yazılar
 Kemik kırığı nasıl hızlı iyileşir? Kırık tedavisinde yeni yöntemler
Kemik kırığı nasıl hızlı iyileşir? Kırık tedavisinde yeni yöntemler- Metabolik kemik hastalıklarında tanı ve tedavi
- Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
- Osteoporoz (kemik erimesi) nedir? Nedenleri, belirtileri ve tedavisi
- Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
 Kemik kırığı nasıl hızlı iyileşir? Kırık tedavisinde yeni yöntemler
Kemik kırığı nasıl hızlı iyileşir? Kırık tedavisinde yeni yöntemler Metabolik kemik hastalıklarında tanı ve tedavi
Metabolik kemik hastalıklarında tanı ve tedavi Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri
Kadınlarda ve erkeklerde en sık görülen kanser türleri ve tedavileri Osteoporoz (kemik erimesi) nedir? Nedenleri, belirtileri ve tedavisi
Osteoporoz (kemik erimesi) nedir? Nedenleri, belirtileri ve tedavisi Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
Çocuk gelişimi ve psikolojisi: Gelişimini etkileyen faktörler
YORUMUNUZ VAR MI?